
Chimica organica
Chimica organica
Appunti, riassunti, nozioni di base, sintesi, schemi, brevi tesine, utili anche agli studenti delle scuole medie, scuole superiori e università
La chimica organica è quel ramo della chimica che studia i composti del carbonio e le loro possibili reazioni. Fino al 1850 molti chimici credevano che i composti inorganici dovessero provenire dal regno minerale mentre quelli organici da organismi viventi e che quest’ ultimi non potessero essere sintetizzati da sostanze inorganiche. Attualmente la divisione tra composti organici e inorganici è convenzionale perché i composti organici possono essere ottenuti in laboratorio.
Fonte: http://galilei2d.altervista.org/wordpress/wp-content/uploads/2008/11/la-chimica-organica.doc
Chimica organica II
ALCHILAZIONE DI CARBONI NUCLEOFILI
Introduzione
I fattori cruciali che devono essere considerati per effettuare un’alchilazione sono:
1) Le condizioni per la generazione di un carbonio nucleofilo
2) L’effetto delle condizioni di reazione sulla struttura e reattività del nucleofilo
3) Regio e stereo selettività della reazione di alchilazione
4) Il ruolo dei solventi, dei controioni e degli altri componenti di reazione che possono influenzare il livello di reazioni collaterali.
1.1. Generazione di carbanioni per deprotonazione
La generazione di carboanioni per deprotonazione richiede una scelta di una appropriata base di Bronsted. L’equilibrio favorirà la formazione dei carboanioni solo quando l’acidità del carbone acido è maggiore di quella dell’acido coniugato della base usata per la deprotonazione. Siccome molti composti carbonilici sono poco acidi (Pka >15) misure accurate della loro acidità sono impossibili in acqua, di conseguenza vengono determinate in solventi organici, e riferite al Pka in modo apprassimativo (consultare tabella 1-1).
I valori di Pk in DMSO sono normalmente più grandi che in acqua perché l’acqua stabilizza gli anioni tramite legami idrogeno più di quanto possa fare il DMSO.
L’ordine dei sostituenti che stabilizzano i carboanioni è:
NO2 > COR > CN » CO2R > SO2R > Ph » SR > H > R
Per ottenere una completa conversione dei chetoni in enolati è necessario usare solventi aprotici, in modo che la deprotonazione del solvente non competa con la formazione dell’enolato.
Basi utilizzate:
NaNH2; base coniugata di MSO; anione trifenilmetile; LDA (litiodiisopropilammide); sali di Li, Na e K di esametildisilazono.
Solventi aprotici utilizzati:
Tetraidrofurano e dimetossi etano(DME)
Altra caratteristica fisica importante è il grado di aggregazione del carbonio, che può essere influenzato sia dal solvente che dal controione.
Vedi Schemi 1.1 e 1.2
1.2. Regioselettività e stereoselettività nella formazione dell’enolato
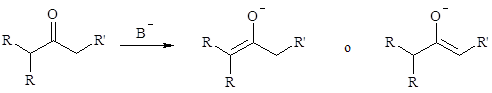 Un chetone dialchilico asimmetrico può formare due enolati regioisomeri per deprotonazione.
Un chetone dialchilico asimmetrico può formare due enolati regioisomeri per deprotonazione.
La composizione di una miscela di enolati può essere governata da fattori cinetici o termodinamici.
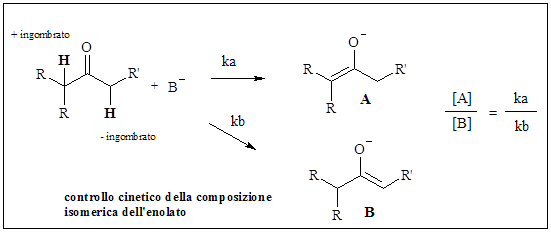 L’enolizzazione è governata da un controllo cinetico quando la deprotonazione è rapida, quantitativa e irreversibile.
L’enolizzazione è governata da un controllo cinetico quando la deprotonazione è rapida, quantitativa e irreversibile.
 E’ invece controllata in modo termodinamico quando i due enolati A e B formati dallo stesso chetone asimmetrico, possono essere interconvertiti; si stabilisce un equilibrio e la composizione dei prodotti riflette la stabilità termodinamica degli enolati (si viene meglio a formare l’enolato più sostituito).
E’ invece controllata in modo termodinamico quando i due enolati A e B formati dallo stesso chetone asimmetrico, possono essere interconvertiti; si stabilisce un equilibrio e la composizione dei prodotti riflette la stabilità termodinamica degli enolati (si viene meglio a formare l’enolato più sostituito).
Il controllo cinetico si può avere usando una base molto forte come LDA o esametildisilammide in un solvente aprotico e in difetto di chetoni.
Il litio è un controione migliore del sodio e del potassio per la generazione regioselettiva di un enolato cinetico. Infatti il litio mantiene un legame migliore con l’ossigeno e riduce così il grado di scambio di protoni.
Solventi aprotici sono indispensabili, perché solventi protici permettono un equilibrio degli enolati dovuto a un giuoco di reversibilità protonazione-deprotonazione che da origine ad una composizione di enolati controllati termodinamicamente.
Le condizioni di controllo cinetico di solito favoriscono l’enolato meno sostituito. Il motivo principale perché si ha questo risultato è perché la rimozione dell’idrogeno meno impedito e più veloce, per ragiono steriche. La rimozione del protone meno impedito conduce all’enolato meno sostituito.
I fattori sterici nella deprotonazione dei chetoni possono essere accentuate usando basi estremamente impedite. La base più largamente usata è lo ione di esametilammide, come sale di litio o di sodio. Altre basi più impedite possono essere usate per questo scopo.
In equilibrio l’enolato più sostituito di solito è la specie dominante. La stabilità del doppio legame C-C aumenta con l’aumento dei sostituenti, e quest’effetto porta alla maggiore stabilità dell’enolato più sostituito.
I termini controllo cinetico e controllo termodinamico sono applicabili alle altre reazioni di formazione di enolati. Possiamo affermare che un reagente o un insieme di condizioni favorisce la “produzione termodinamica“. Questa frase intende che il funzionamento del meccanismo è tale che dopo la formazione i vari possibili prodotti sono all’equilibrio.Quando questo è vero, il prodotto dominante può essere predetto considerando la stabilità relativa dei vari prodotti. Se una reazione è regolata a basso “ controllo cinetico “, la predizione o l’interpretazione delle quantità relative dei prodotti devono essere fatte analizzando le velocità che concorrono alla formazione dei prodotti.
E’ anche possibile realizzare enantioselettività nella formazione dell’enolato usando basi chirali.
 La deprotonazione sotto controllo cinetico di chetoni a,b-insaturi di solito accade preferibilmente al carbonio in alfa adiacente al gruppo carbonilico. L’effetto polare del gruppo carbonilico è probabilmente responsabile per la deprotonazione più veloce a questa posizione.
La deprotonazione sotto controllo cinetico di chetoni a,b-insaturi di solito accade preferibilmente al carbonio in alfa adiacente al gruppo carbonilico. L’effetto polare del gruppo carbonilico è probabilmente responsabile per la deprotonazione più veloce a questa posizione.
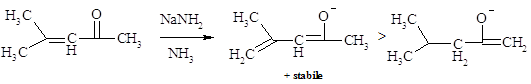 Sotto le condizioni del controllo termodinamico è l’enolato corrispondente alla deprotonazione al carbonio gamma che è presente in maggiore quantità.
Sotto le condizioni del controllo termodinamico è l’enolato corrispondente alla deprotonazione al carbonio gamma che è presente in maggiore quantità.
Il I isomero di enolato si differenzia dal II perché è pienamente coniugato, infatti il sistema p nel II è croce-coniugato. Nell’isomero II, la delocalizzazione della carica negativa è ristretta all’ossigeno e al carbonio in alfa, mentre nel sistema coniugato del I, la carica negativa è delocalizzata sia sull’ossigeno e sul carbonio in alfa sia in quello gamma.
1.3. Altri aspetti della generazione degli enolati
Il raggiungimento di condizioni sotto le quali i litio enolati sono stabili e non equilibrano con i regioisomeri, permette l’uso di altre reazioni in addizione all’eliminazione del protone. Ad esempio per generare enolati specifici.
Alcuni metodi sono mostrati nello Schema 1.4.
Il cloruro del trimetilsilil metano è adoperato appunto per formare il trimetilsilil etere dell’enolo. In questo modo si può in caso di una miscela separare i diversi regioisomeri per via cromatografica (per i sali, e gli enolati di litio lo sono, sarebbe molto dispendiosa una separazione).
Questi composti una volta formati possono essere lavati (cleavage) tramite metil litio (esempi 1 e 3 schema 1.4) o sali di tetraalchilammonio fluoruro (esempio 2).
La forza che guida quest’ultima tecnica di lavaggio è la formazione di un legame molto forte Si-F (142 kcal/mol).
Il trimetil silil enol etere può essere preparato direttamente dai chetoni. Una procedura include la reazione con il trimetil silil cloruro ed un’ammina terziaria. In questo modo si può ottenere enol etere termodinamicamente più stabile.
La stessa cosa vale per il t-butildimetil silil cloruro con il potassio idruro come base. Il trimetilsilil trifluorometano solfonato, il quale è più reattivo, da principalmente il prodotto meno sostituito.
Alte rese di trimetilsililenoletere meno sostituito possono essere inoltre raggiunte adoperando in una miscela di chetone e trimetilsilil cloruro le condizioni adatte per un controllo cinetico (- 78°C e LDA come base). Il metodo si può ulteriormente raffinare utilizzando basi più ingombranti come la t-octil-t-butilammina.
Ancora un altro metodo per preparare trimetil silil enoleteri specifici può risultare la riduzione 1,4 di chetoni a-b insaturi usando silani come riducenti ed un complesso Pt – diviniltetrametildisilossano come catalizzante (esempio 7), oppure Li e NH3 (esempio 6). Altri metodi verranno discussi più avanti.
1.4. Alchilazione degli enolati
L’alchilazione degli enolati è un processo sintetico molto importante. L’alchilazione di composti relativamente acidi come b-dichetoni, b-chetoesteri e esteri di acidi malonici può essere effettuata usando alcol come solventi e metalli alcossidi come basi.
La presenza di due gruppi sostituenti elettronattrattori facilita la formazione di un enolato risultante dalla rimozione di un protone dall’atomo di carbonio situato tra loro. L’alchilazione quindi va avanti con un processo SN2.
Alcune reazioni sono mostrate nello Schema 1.5.
L’agente alchilante deve essere molto reattivo nei confronti del posizionamento della carica nucleofila.
Alogenuri primari o solfonati, specialmente l’allilico ed il benzilico, sono gli agenti alchilanti migliori.
Sistemi secondari reagiscono molto lentamente e spesso danno rese basse per reazioni competitive di eliminazione. Alogenuri terziari danno solo prodotti di eliminazione.
Il gruppo metilenico acido può essere dialchilato se l’agente alchilante e la base sono in quantità sufficienti.
La dialchilazione può essere però un aspetto indesiderato della reazione se si vuole ottenere un prodotto monoalchilato.
L’uso di dialogenoalcani come agenti alchilanti porta alla formazione di anelli (esempio 7), si ha quindi la sintesi di composti ciclici per alchilazione intramolecolare.
I carboni relativamente acidi come quelli degli esteri malonici e dei b-chetoesteri furono la prima classe di composti utilizzata per produrre carbanioni per il semplice fatto che essi deprotonano facilmente tramite l’uso di ioni alcossido (i quali sono agevolmente preparabili).
La preparazione dei 2-sostituiti b-chetoesteri (esempi 1,4 e 8 sch.1.5) e dei 2-sostituiti derivati dell’estere malonico (esempi 2 e 7) è utile anche per la sintesi di chetoni e di acidi carbossilici (vedi sintesi acetoacetica dei chetoni). Infatti, sia i b-chetoacidi sia gli acidi malonici vanno incontro a facile decarbossilazione.
I carbanioni malonato ed acetoacetato sono gli equivalenti sintetici di carbanioni sintetici privi dei sostituenti degli esteri.Nella preparazione del 2-eptanone (es. 1, sch. 1.5 e 1.6), l’etilacetoacetato funge da equivalente sintetico dell’acetone. E’ anche possibile usare il derivato dilitio dell’acido acetoacetico come equivalente sintetico dell’enolato acetone.
In questo caso la decarbossilazione può verificarsi direttamente sul prodotto di alchilazione.
L’uso dei b-chetoesteri e degli esteri malonici è stato largamente soppiantato dallo sviluppo di più nuove procedure basate sulla formazione selettiva di enolati. Queste procedure permettono la diretta alchilazione del chetone e degli enolati degli esteri evitando l’idrolisi e la decarbossilazione dei chetoesteri intermedi.
La maggior parte delle alchilazioni sono estrapolate dalla deprotonazione del chetone sotto controllo cinetico o termodinamico. Altri metodi di preparazione degli enolati sono stati già affrontati nella Sezione 1.3 e alcuni esempi dell’alchilazione degli enolati del chetone sono dati nello Schema 1.7.
Un aspetto della reazione che è cruciale in molti casi è la stereoselettività. Lo step dell’alchilazione ha una preferenza stereoelettronica per l’approccio dell’elettrofilo perpendicolarmente al piano dell’enolato, in modo che gli elettroni coinvolti nella formazione del legame sono elettroni p.
L’elettrofilo attaccherà la meno ingombrata delle due facce, ed il livello di stereoselettività dipenderà solo dal differente ingombro sterico.
Ad esempio, il 4-t-butilcicloesanone potrà essere attaccato da un alchilante da entrambe le facce del piano dell’enolato, poiché l’opposizione sterica incontrata durante l’approccio dell’elettrofilo è minima.
Di conseguenza il rapporto fra i due steroisomeri alchilati sarà cis/trans = 1/1.


Il prodotto cis si può formare con uno stato di transizione che ha una conformazione twist atta a raggiungere i requisiti per un controllo stereoelettronico. Il fatto che questo percorso non è sfavorito è coerente con l’altra prova che lo stato di transizione nell’alchilazione dell’enolato si raggiunge presto e riflette principalmente le caratteristiche di struttura del reagente, non del prodotto. Uno stato di transizione tardivo potrebbe sfavorire la formazione dell’isomero cis per l’energia di tensione associata alla conformazione non chair del prodotto.
 L’introduzione di sostituenti alchilici al carbonio in a nell’enolato intensifica la steroselettività in qualche modo. Per minimizzare l’interazione sterica con l’ossigeno solvatato, il gruppo alchilico è distorto dalla coplanarità. Ciò previene l’enolato dall’attacco dell’elettrofilo dalla direzione assiale. L’approccio alternativo dalla faccia superiore aumenterebbe l’interazione sterica forzando il gruppo alchilico a eclissarsi con l’ossigeno dell’enolato.
L’introduzione di sostituenti alchilici al carbonio in a nell’enolato intensifica la steroselettività in qualche modo. Per minimizzare l’interazione sterica con l’ossigeno solvatato, il gruppo alchilico è distorto dalla coplanarità. Ciò previene l’enolato dall’attacco dell’elettrofilo dalla direzione assiale. L’approccio alternativo dalla faccia superiore aumenterebbe l’interazione sterica forzando il gruppo alchilico a eclissarsi con l’ossigeno dell’enolato.
Quando un metile addizionale è piazzato al C-3, c’è una forte preferenza per l’alchilazione anti al gruppo 3-metile. Questo può essere attribuito alla conformazione dell’enolato, che piazza il metile in una conformazione pseudoassiale a causa di una tensione allilica (vedi parte A sez. 3.3).
Il gruppo 3-metile quindi protegge la parte più bassa dell’enolato.

Se l’alchilazione è intramolecolare, le restrizioni addizionali di conformazione riguardo l’approccio dell’elettrofilo sull’enolato diventano importanti.
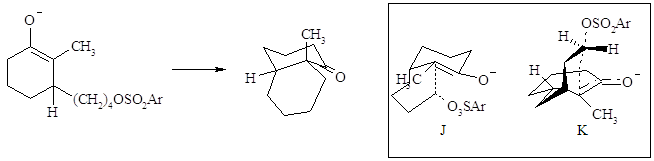
I principi generali che regolano gli stadi di energia della chiusura intramolecolare per formare una struttura ciclica sono stati riassunti da Baldwin (vedi Parte A, Sez. 3.9).
Lo stato di transizione deve avere una geometria tale da permettere l’interazione degli orbitali p dell’enolato, in modo da raggiungere approssimativamente un allineamento con il gruppo uscente.
L’alchilazione probabilmente procede attraverso uno stato di transizione J. Lo stato di transizione K per la formazione dell’anello a giunzione trans sarebbe maggiormente teso per la necessità di attraversare la faccia opposta del sistema p dell’enolato.
Ricapitolando i maggiori fattori che contribuiscono alla stereoselettività dell’alchilazione dell’enolato sono:
- conformazione dell’enolato
- requisiti stereoelettronici per permettere un approccio a traiettoria approssimativamente perpendicolare
- preferenza sterica per il meno ingombrato percorso di approccio elettrofilo.
- Generazione e dialchilazione di dianioni
In presenza di basi sufficientemente forti, come un alchil litio, sodio alchile, potassio idruro, sodio o potassio amide, o LDA, i composti 1-3 dicarbonilici possono essere convertiti nei loro dianioni attraverso due deprotonazioni sequenziali.

E’ importante e da tener presente che:
le reazioni di alchilazione dei dianioni avvengono sul carbonio più basico e non sul metilene attivato che si trova fra i due carbonili (come avveniva nell’alchilazione del mono-enolato).
Quindi secondo la forza e la quantità della base si possono ottenere composti 1-3 dicarbonilici alchilati sul metilene attivato o sul carbonio meno acido.
Una serie di esempi sono mostrati nello Schema 1.8.
- Effetti del solvente e del controione nell’alchilazione degli enolati
Il tasso di alchilazione degli ioni enolati è fortemente dipendente dal solvente nel quale la reazione è portata avanti.
DMSO e N,N-dimetilformammide (DMF) sono particolarmente effettivi nell’incoraggiare la reattività degli ioni enolato.
Questi due composti appartengono alla classe dei solventi polari aprotici. Altri membri di questa classe sono l’N-metilpirrolidone (NMP) e la esametil triammide fosforica (HMPA).
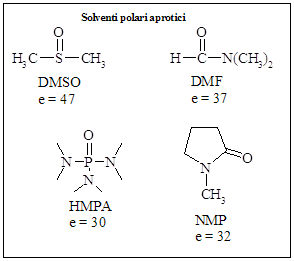 I solventi polari aprotici sono composti che hanno un’alta costante dielettrica ma che mancano di gruppi idrossilici o altri gruppi con legami a idrogeno. Possiedono inoltre un’alta abilità nel coordinare i cationi metallici, in modo che loro possano solvatare e dissociare gli enolati ed altri carbanioni da coppie ioniche e gruppi.
I solventi polari aprotici sono composti che hanno un’alta costante dielettrica ma che mancano di gruppi idrossilici o altri gruppi con legami a idrogeno. Possiedono inoltre un’alta abilità nel coordinare i cationi metallici, in modo che loro possano solvatare e dissociare gli enolati ed altri carbanioni da coppie ioniche e gruppi.
Il più alto livello di reattività, il quale può essere avvicinato ma non raggiunto in soluzione, è quello del “nudo” anione enolato desolvatato.
Per una coppia ionica enolato-ione metallico in soluzione, la massima reattività la si potrebbe aspettare in un intermedio nel quale il catione sia prevalentemente solvatato rispetto all’anione enolato che invece deve essere il meno solvatato possibile.
I solventi polari aprotici sono buoni solvatatori di cationi e scarsi solvatatori di anioni.
Ciò è assimilabile al fatto che ciascuno di essi ha un O polarizzato negativamente, avviabile per la coordinazione al catione metallo-alcalino.

Perciò questi solventi danno un intermedio nel quale la coppia ionica metallo-enolato viene dissociata per dare un anione enolato meno ingombrato e più reattivo.
I solventi polari protici possiedono anch’essi un’abilità pronunciata di separare le coppie ioniche ma sono meno favoriti come solventi per l’alchilazione degli enolati perché loro coordinano insieme sia lo ione metallico, sia lo ione enolato.
Avviene così la solvatazione dell’anione enolato mediante legami a idrogeno. L’anione enolato solvatato può reagire così in tutt’altro modo e determinare una O-alchilazione al posto di una C-alchilazione.

Il THF e il DME sono solventi leggermente polari e sono dei moderati solvatatori di cationi.
La coordinazione al catione metallico coinvolge la coppia solitaria dell’atomo di O.
Questi solventi, per la loro bassa costante dielettrica, sono meno effettivi nel separare le coppie ioniche o gli aggregati rispetto ai solventi polari aprotici.
Le strutture cristalline del litio e potassio enolato del metil t-butil chetone sono state determinate grazie alla cristallografia a raggi X e sono rappresentate nelle figure (per questo motivo si parla di “aggregati”).
La figura rappresenta lo stato solido, invece aggregati esamerici costituiscono una buona indicazione della natura degli enolati in solventi debolmente coordinanti.
Nonostante tutto, il THF ed il DME sono i solventi più comunemente utilizzati per alchilazioni di enolati.
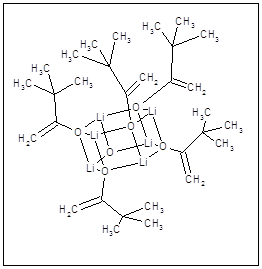
Essi inoltre sono i solventi maggiormente consigliati per una generazione di enolati che devono essere alchilati sotto controllo cinetico e presentano dei vantaggi nelle fasi della purificazione e del “workup” sui prodotti rispetto ai solventi polari aprotici. La reattività degli enolati in questi solventi può essere rafforzata aggiungendo un reagente che può legare i cationi metallo-alcalini con più forza.
Esempi di questi reagenti sono il tetrametiletilendiammina (TMEDA) e gli eteri ciclici.
La reattività degli enolati è anche influenzata dal tipo di controione adoperato nella base.
Tra gli ioni più comunemente usati l’ordine di reattività è:
Mg2+ < Li+ < Na+ < K+
Il motivo di quest’ordine di reattività è riconducibile alle caratteristiche dei solventi. I cationi più piccoli e con una carica più densa, Mg2+ e Li+, sono maggiormente associati all’enolato rispetto agli ioni Na+ e K+.
- Ossigeno o Carbonio come sito di alchilazione
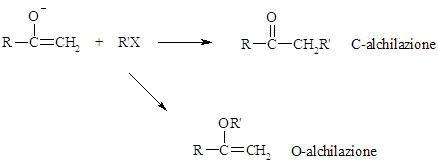 Gli anioni enolati sono nucleofili ambidentati. L’alchilazione di un enolato può avvenire sia al carbonio che all’ossigeno. Considerando che la carica negativa dovrebbe risiedere sull’O, la O-alchilazione dovrebbe essere dominante. E’ possibile però stabilire delle condizioni di reazione tali da favorire l’alchilazione sul carbonio.
Gli anioni enolati sono nucleofili ambidentati. L’alchilazione di un enolato può avvenire sia al carbonio che all’ossigeno. Considerando che la carica negativa dovrebbe risiedere sull’O, la O-alchilazione dovrebbe essere dominante. E’ possibile però stabilire delle condizioni di reazione tali da favorire l’alchilazione sul carbonio.
La O-alchilazione è più pronunciata quando l’enolato è dissociato. Quindi essa è comandata sia dalla natura del solvente, che da quella del controione.
Nel momento in cui il sale potassico del etil acetoacetato è trattato con dietil solfato nel solvente polare aprotico HMPA, il prodotto maggiore (83%) è l’O-alchilato.
Nel THF, dove avviene l’aggruppamento degli ioni, tutto il prodotto è C-alchilato. Nel t-butanolo, dove l’anione acetoacetato è legato con un ponte a idrogeno dal solvente, si osserva ancora la C-alchilazione.
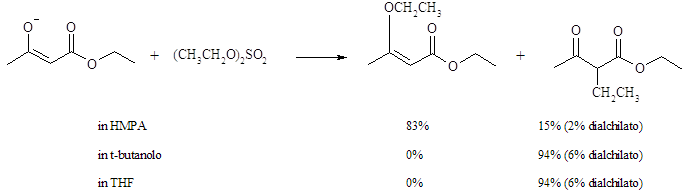
Rapporti più alti della C/O alchilazione sono osservati con gli alogenuri alchilici, piuttosto che con i solfonati ed i solfati. Il più alto fra questi è quello osservato per lo ioduro alchilico. Per l’etilazione del sale potassico dell’etile acetoacetato nell’HMPA, vengono ottenute le composizioni del prodotto mostrate sotto.

Gli effetti del gruppo uscente sul rapporto C/O alchilazione può essere correlato dalla corrispondenza
“hard-soft-acid-base”. Dei due siti nucleofili di uno ione enolato, l’O è più “hard” del C.
Le reazioni di sostituzione nucleofila di tipo SN2 procedono meglio quando sia il nucleofilo che il gruppo uscente sono entrambi o hard o soft.
Di conseguenza, lo ioduro d’etile, con il gruppo uscente molto soft quale lo iodio, da l’alchilazione al carbonio piuttosto che all’ossigeno.
D’altra parte, gruppi uscenti con l’O, come i solfonati o i solfati , sono gruppi hard e quindi l’alchilazione si ha una O-alchilazione.
La combinazione hard-hard è anche favorita da uno stato di transizione precoce, dove la distribuzione di cariche è un fattore molto importante.
La combinazione soft-soft invece è favorita da un tardo stato di transizione, dove la parziale formazione del legame è il fattore dominante.
In definitiva il prodotto della C-alchilazione è più stabile di quello della O-alchilazione poiché l’energia di legame di C=O + C-C è maggiore dell’energia di legame C=C + C-O.
Perciò le condizioni che favoriscono un dissociato, e più reattivo enolato, favoriscono la O-alchilazione.
Per riassumere: la resa per la O-alchilazione è massima dall’uso di solfati alchilici o solfati arilici in un solvente polare aprotico;
la resa per la C-alchilazione è massima se si usa un alogenuro alchilico in un solvente meno polare o protico.
L’alchilazione intramolecolare degli enolati conduce alla formazione di prodotti ciclici.
In addizione agli altri fattori che governano l’equilibrio C/O nelle rese dell’alchilazione, l’elemento del controllo stereoelettronico gioca un ruolo importante in casi del genere.
Per far si che la C-alchilazione possa avvenire, l’orbitale p del carbonio in a deve essere allineato con il legame C-Br nella geometria lineare associata con lo stato di transizione SN2. Quando l’anello da chiudere è a sei membri, questa geometria è accessibile, e la ciclizzazione a cicloesanone si realizza. Con anelli a cinque membri, l’allineamento non può essere raggiunto facilmente.
La ciclizzazione all’O avviene quindi più facilmente della formazione del ciclopentanone. Lo stato di transizione per la O-alchilazione coinvolge un orbitale dell’O con una coppia solitaria di elettroni (lone-pair orbital) e porta ad una minore tensione rispetto allo stato di transizione della C-alchilazione.

Negli enolati formati dall’eliminazione di un protone nei chetoni a-b insaturi, ci sono tre siti potenzialmente adatti per un’alchilazione o per una protonazione: l’O, l’a-C ed il g-C. Il sito preferito cineticamente è l’a-C.
La protonazione degli enolati è un utile metodo per trasformare composti a-b insaturi (chetoni, esteri) nei corrispettivi e meno stabili b-g insaturi.

Un caso interessante da analizzare è quello degli ioni fenossido; essi danno solitamente la O-alchilazione poiché la C-alchilazione dovrebbe rompere la coniugazione aromatica dell’anello benzenico.
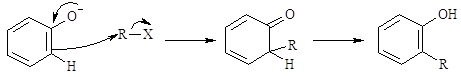
 Tuttavia in solventi che possono stabilire con l’O dei legami idrogeno molto forti, si possono misurare alte rese di C-alchilazione.
Tuttavia in solventi che possono stabilire con l’O dei legami idrogeno molto forti, si possono misurare alte rese di C-alchilazione.
Fonte:http://digilander.libero.it/ctfonline/appunti/file/organica2.doc
Chimica organica
CHIMICA ORGANICA : è la chimica dei composti del carbonio,
(eccetto l’ossido di carbonio, l’anidride carbonica, l’acido carbonico e i carbonati)
I più semplici composti organici sono gli idrocarburi, composti di carbonio e idrogeno;
tra loro quello a molecola più piccola è il metano, CH4 , il gas che si usa in cucina;
la benzina è una miscela di idrocarburi.
I legami tra carbonio e carbonio possono essere singoli, doppi o tripli.
Se una molecola contiene solo legami singoli C-C, è detta satura (di idrogeno);
se contiene anche un solo legame doppio o triplo è detta insatura;
le sostanze insature possono addizionare idrogeno (idrogenazione), rompendo i legami multipli, e diventare così sature; ad esempio, dall’idrogenazione di oli vegetali (insaturi) si può ottenere la margarina.
Lo “scheletro carbonioso” può essere una catena più o meno lunga, lineare o ramificata, o una struttura ciclica, cioè chiusa ad anello.
Un anello particolare è quello dei composti aromatici: è esagonale, con legami singoli e doppi alternati; si trova nel benzene ( un idrocarburo aromatico con molecola esagonale molto stabile, cancerogeno), ma anche nella naftalina, nell’acido acetil salicilico (è il principio attivo dell’Aspirina), e nell’adrenalina.
A volte ad una stessa formula bruta possono corrispondere due o più formule di struttura diverse; i composti che hanno la stessa formula bruta ma diverse formule di struttura vengono detti isomeri.
Le proprietà delle varie classi di composti organici sono determinate dai GRUPPI FUNZIONALI: si tratta di piccoli raggruppamenti di atomi che sono in grado di conferire un particolare comportamento chimico ad una molecola.
I principali sono riassunti nello schema sottostante, insieme ad altri esempi di composti organici.
- –OH gruppo ossidrile:
è polare, idrofilo, può formare legami idrogeno.
Si trova nei seguenti gruppi di sostanze:
alcoli, come l’alcol metilico o metanolo CH3OH, sostanza molto tossica, usato come combustibile e solvente, l’ alcol etilico o etanolo CH3CH2OH, meno tossico del precedente, contenuto nel vino, nella birra, nei superalcolici, e utilizzato anche per disinfettare (alcol denaturato) o come solvente.
zuccheri (es: glucosio C6H12O6)
- - COOH gruppo carbossilico:
è polare, si comporta da acido debole (tende a perdere H+)
Si trova, ad esempio, nei seguenti gruppi di sostanze:
acidi carbossilici come l’acido formico HCOOH, l’acido acetico CH3COOH, l’acido lattico CH3CHOHCOOH
acidi grassi , composti da catena carboniosa lineare contenente da 4 a 24 atomi di Carbonio (generalmente i C sono in numero pari), come l’acido laurico, palmitico, stearico (saturi), o l’acido oleico, linoleico, arachidonico, gli omega 3 (insaturi).
amminoacidi.
- -NH2 gruppo amminico:
è polare, si comporta da base debole (accetta protoni);
si trova, ad esempio, negli amminoacidi e nell’urea
- =CO gruppo carbonilico
è polare;
Si trova, ad esempio, nei seguenti gruppi di sostanze:
aldeidi (in cui si può trovare solo all’inizio o alla fine della molecola), come la formaldeide CH2O, l’acetaldeide, e i costituenti degli oli essenziali di lavanda, citronella, eucalipto.
chetoni (in cui non si trova mai in posizione iniziale o terminale), come l’acetone CH3COCH3, e la canfora, sostanze piuttosto volatili spesso usate come aromi artificiali, in profumeria o come solventi
zuccheri.
- -CH3 gruppo metilico:
è apolare, idrofobo
- gruppo fosfato
è polare, acido (deriva dall’acido fosforico H3PO4)
Si trova, ad esempio, nei seguenti gruppi di sostanze:
nucleotidi e derivati, come DNA, RNA, ATP
fosfolipidi
fonte: http://digilander.libero.it/bionote2008/organica.doc
Chimica organica
GLOSSARIO DI CHIMICA ORGANICA
Acetale: Composto organico in cui ad uno stesso atomo di carbonio sono legati due gruppi alcossile o due gruppi fenossile o un alcossile e un fenossile
Achirale: Privo di chiralità; una molecola è achirale se possiede un piano di simmetria e quindi è sovrapponibile alla propria immagine speculare
Acido carbossilico: Composto organico contenente il gruppo funzionale -COOH
Acido coniugato: Prodotto risultante dalla protonazione di una base di Brønsted-Lowry
Acido di Arrhenius: Sostanza che si ionizza in soluzione acquosa liberando idrogenioni H+
Acido di Brønsted-Lowry: Sostanza in grado di donare uno ione idrogeno ad una base
Acido di Lewis: Sostanza con un orbitale vuoto a bassa energia che può accettare una coppia di elettroni da una base per formare un nuovo legame covalente
Acido solfonico: acido organico che ha la struttura generale RSO3H o ArSO3H
Acilazione di Friedel-Crafts Reazione di sostituzione elettrofila aromatica che introduce un gruppo acilico nell’anello aromatico; si effettua su un substrato aromatico mediante un alogenuro acilico (o un’anidride) usando un acido di Lewis come catalizzatore
Addizione 1,2: Addizione di un reagente alle due estremità di un doppio legame
Addizione 1,4: Addizione di un reagente alle estremità di un sistema p coniugato; i dieni coniugati danno addotti 1,4 se trattati con elettrofili come HCl. I composti carbonilici a-b insaturi danno addotti 1,4 se trattati con nuclofili come lo ione cianuro
Addizione anti: Addizione di atomi o gruppi di atomi che avviene dal lato o dalla faccia opposta di un legame p. Nei sistemi ciclici l’addizione anti è equivalente ad un’addizione trans.
Addizione coniugata: Addizione di un nucleofilo all’atomo di carbonio b di un composto carbonilico a-b insaturo
Addizione elettrofila: reazione di addizione in cui il primo stadio del meccanismo implica un’addizione del termine elettrofilo del reagente a un doppio legame carbonio-carbonio
Addizione nucleofila: Reazione tipica dei sistemi carbonilici in cui un nucleofilo attacca il carbonio del C=O aprendo il doppio legame e all’ossigeno si lega un idrogenione
Addizione sin: Addizione di atomi o di gruppi di atomi allo stesso lato od alla stessa faccia di un legame p
Alcano: Idrocarburo alifatico avente solo legami C-H e C-C di tipo s
Alcano a catena ramificata: Alcano contenente uno o più sostituenti alchilici legati alla catena carboniosa principale
Alcano normale: Alcano a catena lineare, opposto di un alcano ramificato
Alchene: Idrocarburo contenente un doppio legame C=C
Alchene disostituito: Alchene caratterizzato da due gruppi alchilici e due idrogeni legati ai carboni del doppio legame
Alchene interno: Alchene che possiede almeno un atomo di carbonio legato a ciascun atomo del doppio legame
Alchene monosostituito: Alchene che contiene un solo gruppo alchilico e tre atomi di idrogeno legati ai due atomi di carbonio che formano il doppio legame
Alchene terminale: Alchene in cui il doppio legame si trova all’estremità della catena degli atomi di carbonio
Alchene tetrasostituito: Alchene che contiene quattro gruppi alchilici e nessun atomo di idrogeno legato agli atomi di carbonio del doppio legame
Alchene trisostituito: Alchene che contiene tre gruppi alchilici e un atomo di idrogeno direttamente legati ai carboni del doppio legame
Alchilazione: Reazione che trasferisce un gruppo alchilico da un atomo ad un altro; alchilazione di Friedel-Craft di anelli aromatici, alchilazione di un’ammina, sintesi di Williamson di eteri ecc.
Alchino: Idrocarburo che contiene un triplo legame C≡C
Alchino interno: Un alchino che ha un atomo di carbonio legato a ciascuna estremità del triplo legame
Alchino terminale: Alchino in cui il triplo legame si trova all’estremità della catena degli atomi di carbonio
Alcool: Composto organico che contiene un gruppo ossidrile (OH) legato ad un atomo di carbonio ibridato sp3
Alcool primario: Alcool, avente la struttura generale RCH2OH, nel quale l’atomo di carbonio legato al gruppo ossidrile è legato a un solo atomo di carbonio
Alcool secondario: Alcool, avente la struttura generale R2CHOH, nel quale l’atomo di carbonio legato al gruppo ossidrile è legato a due atomi di carbonio
Alcool terziario: Alcool, avente la struttura generale R3COH, nel quale l’atomo di carbonio legato al gruppo ossidrile è legato a tre atomi di carbonio
Alcossido: Anione all’ossigeno, nucleofilo e basico, formato dalla deprotonazione di un alcool con una base. Gli alcossidi hanno la struttura RO-
Aldeide: Composto organico contenente un gruppo carbonilico con un atomo di idrogeno legato all’atomo di carbonio carbonilico (H-C=O)
Aliciclico: Idrocarburo alifatico ciclico, come un cicloalcano o un cicloalchene
Alifatico: Composto, o parte di un composto, in cui compaiono legami carbonio-carbonio di tipo s e p (alcani, alcheni o alchini), ma non gruppi aromatici
Allile: Sostituente avente la struttura –CH2-CH=CH2
Allilico: Termine usato per indicare la posizione successiva ad un doppio legame carbonio-carbonio. L’atomo di carbonio legato a quello di un doppio legame carbonio-carbonio è un atomo di carbonio allilico e un sostituente legato ad un carbonio allilico è un sostituente allilico
Alogenazione: Reazione di un composto organico con alogeni
Alogenuro acilico: Composto avente formula generale RCOX dove R è un gruppo alchilico o acrilico e X è un alogeno
Alogenuro alchilico: Molecola organica contenente un atomo di alogeno legato a un atomo di carbonio ibridato sp3
Alogenuro alchilico primario: Alogenuro alchilico avente la struttura generale RCH2X, nel quale l’atomo di carbonio legato all’alogeno è legato a un solo atomo di carbonio
Alogenuro alchilico secondario: Alogenuro alchilico avente la struttura generale R2CHX, nel quale l’atomo di carbonio legato all’alogeno è legato a due atomi di carbonio
Alogenuro alchilico terziario: Alogenuro alchilico avente la struttura generale R3CX, nel quale l’atomo di carbonio legato all’alogeno è legato a tre atomi di carbonio
Alogenuro allilico: Molecola organica contenente un atomo di alogeno legato al carbonio adiacente a un doppio legame carbonio-carbonio
Alogenuro arilico: Molecola organica contenente un atomo di alogeno legato a un anello aromatico
Alogenuro benzilico: Composto aromatico con un atomo di alogeno legato a un atomo di carbonio che è a sua volta legato all’anello benzenico
Alogenuro vinilico: Molecola contenente un atomo di alogeno legato a un carbonio ibridato sp2 di un doppio legame
Aloidrina: Composto caratterizzato da un gruppo ossidrilico e un atomo di alogeno su due atomi di carbonio adiacenti
Ammide: Classe di composti organici contenenti il gruppo funzionale O=CNR2, dove gli R possono essere idrogeno, gruppi alchilici o acrilici o una loro combinazione
Ammide primaria: Un’ammide nella quale l’atomo di azoto legato al carbonio carbonilico ha legati due atomi di idrogeno (RCONH2)
Ammide secondaria: Un’ammide nella quale l’atomo di azoto legato al carbonio carbonilico ha legati un atomo di idrogeno e un gruppo alchilico o arilico (RCONHR)
Ammide terziaria: Un’ammide nella quale l’atomo di azoto legato al carbonio carbonilico ha legati due gruppi alchilici o arilici o una loro combinazione (RCONR2)
Ammina: Composto contenente uno o più sostituenti organici legati a un atomo di azoto, RNH2, R2NH o R3N. Un’ammina possiede un doppietto di non-legame sull’azoto
Ammina primaria: Ammina contenente un atomo di azoto legato a un gruppo R e a due atomi di idrogeno (RNH2)
Ammina secondaria: Ammina contenente un atomo di azoto legato a due gruppi R e a un atomo di idrogeno (R2NH)
Ammina terziaria: Ammina contenente un atomo di azoto legato a tre gruppi R (R3N)
Amminazione riduttiva: Formazione di un’immina da un’aldeide o un chetone seguita dalla sua riduzione ad ammina
Ammino (gruppo): Sostituente contenente azoto avente la struttura –NH2
Anelli fusi: Un sistema biciclico o policiclico in cui due o più anelli condividono un legame tra due atomi adiacenti
Anfotero: Capace di agire sia da acido sia da base
Angolo diedro: L’angolo creato da due piani che si intersecano
Angolo di legame: Angolo formato da due legami adiacenti
Anidride: Composto organico caratterizzato da due gruppi carbonilici uniti da un atomo di ossigeno
Anidride mista: Anidride che ha due gruppi alchilici o arilici diversi legati ai due carboni carbonilici
Anidride simmetrica: Anidride che ha due gruppi alchilici o arilici uguali legati ai due carboni carbonilici
Anione: Ione carico negativamente ottenuto da un atomo neutro quando acquista un elettrone
Anione acetiluro : Anione nucleofilo formato mediante trattamento di un alchino terminale con una base forte (R‑C≡C-)
Anione carbossilato: Anione avente la struttura generale RCOO-, originato dalla deprotonazione di un acido carbossilico con una base di Brønsted-Lowry
Anione enolato: Anione con carica delocalizzata che si ottiene per rimozione di un idrogenione da un enolo o dal composto carbonilico in equilibrio con l’enolo
Anione solfonato: Anione avente la struttura generale RSO3-, formato dalla deprotonazione di un acido solfonico per azione di una base di Brønsted-Lowry
Antiossidante: Composto che impedisce una reazione di ossidazione
Arene: Benzene alchil-sostituito
Arilammina: Composto aromatico ammino-sostituito (ArNH2)
Arile: Sostituente che si ottiene dalla rimozione formale di un idrogeno da un anello aromatico
Aromaticità: Caratteristica speciale di alcune molecole che sono planari, cicliche, posseggono una nube circolare ininterrotta di elettroni p sopra e sotto il piano della molecola, contenente 4n+2 elettroni, e che danno più facilmente reazioni di sostituzione che di addizione
Attacco da retro: L’avvicinarsi del gruppo entrante dalla parte opposta rispetto al gruppo uscente
Attacco frontale: Attacco del gruppo entrante dalla stessa parte del gruppo uscente
Attività ottica: Proprietà di quelle sostanze che fanno ruotare il piano di vibrazione della luce polarizzata
Azocomposto: Composto di struttura generale R-N=N-R
Base coniugata: Anione risultante dalla deprotonazione di un acido di Brønsted-Lowry
Base di Arrhenius: Sostanza che si ionizza in soluzione acquosa liberando ossidrilioni OH-
Base di Brønsted-Lowry: Un composto in grado di accettare protoni. Una base di Brønsted-Lowry deve essere in grado di formare un legame con un protone.
Base di Lewis: Una specie che dona una coppia di elettroni ad un acido; la coppia di elettroni non viene rimossa, ma condivisa con un altro atomo per formare il legame covalente
Base di Schiff: Un composto organico di struttura generale R2C=NR’, viene chiamata anche immina
Benzile: Sostituente contenente l’anello benzenico legato al gruppo CH2 (C6H5CH2-)
Benzilico: Termine che indica un atomo di carbonio legato a un anello benzenico
Benzoile: Sostituente avente la struttura –COC6H5
Bis-ossidrilazione: Reazione di ossidazione in cui due gruppi ossidrilici sono aggiunti a un doppio legame per formare un 1,2-diolo o glicol
Bis-ossidrilazione anti: Reazione di ossidazione che implica l’addizione di due gruppi ossidrilici da facce opposte di un doppio legame
Bis-ossidrilazione sin: Reazione di ossidazione che implica l’addizione di due gruppi ossidrilici dalla stessa faccia di un doppio legame
Bontà di un gruppo uscente: Misura di quanto facilmente un gruppo uscente (Z) può accettare il doppietto elettronico del legame C-Z durante una reazione di sostituzione o di eliminazione
Bromidrina: Composto caratterizzato da un atomo di bromo e un gruppo ossidrilico su atomi di carbonio adiacenti
Bromurazione: introduzione di uno o più atomi di bromo nella molecola di un composto chimico
Cahn-Ingold-Prelog: Studiosi che hanno elaborato il sistema di nomenclatura per designare un centro stereogenico come R o S a seconda della disposizione tridimensionale dei quattro gruppi ad esso legati
Calore di idrogenazione: Quantità di calore (DH°) o entalpia rilasciata quando un doppio legame C=C viene idrogenato
Calore di combustione: Quantità di calore (DH°) svolto da una mole di un composto quando reagisce con ossigeno e gli atomi che lo costituiscono si trasformano nei rispettivi ossidi
Calore di reazione: Energia assorbita o rilasciata in una reazione; è chiamata anche variazione di entalpia (DH°)
Carbanione: Specie ionica contenente un atomo di carbonio trivalente e carico negativamente (R3C:-).
Carbinolammina: Intermedio instabile avente un gruppo ossidrilico e un gruppo amminico sullo stesso atomo di carbonio [R2C(OH)NH2]. La carbinolammina si forma durante la reazione di addizione di un’ammina a un gruppo carbonilico
Carbocatione: Gruppo di atomi contenente un carbonio con soli sei elettroni, quindi con una carica positiva (R3C+)
Carbocatione allilico: Carbocatione che ha la carica positiva su un atomo adiacente a un doppio legame C=C. Un carbocatione allilico è stabilizzato per risonanza
Carbocatione primario: Carbocatione nel quale la carica positiva si trova su un carbonio a sua volta legato ad un solo atomo di carbonio ( Es. RCH2+)
Carbocatione secondario: Carbocatione nel quale la carica positiva si trova su un carbonio a sua volta legato a due atomi di carbonio ( Es. R2CH+)
Carbocatione terziario: Carbocatione nel quale la carica positiva si trova su un carbonio a sua volta legato a tre atomi di carbonio ( Es. R3C+)
Carbonile: Gruppo funzionale che contiene un doppio legame carbonio-ossigeno (C=O)
Carbonio a: Carbonio adiacente a un gruppo funzionale. Per esempio: in una reazione di eliminazione, il carbonio che è legato al gruppo uscente; in un composto carbonilico, il carbonio che è legato al carbonio carbonilico
Carbonio allilico: Atomo di carbonio adiacente all’atomo di carbonio di un doppio legame C=C
Carbonio asimmetrico: Atomo di carbonio che lega quattro gruppi differenti
Carbonio b: Carbonio successivo a quello adiacente a un gruppo funzionale. Per esempio: in una reazione di eliminazione, il carbonio adiacente al carbonio che è legato al gruppo uscente; in un composto carbonilico, il carbonio che è distante due atomi di carbonio dal carbonio carbonilico
Carbonio primario: In una molecola, un atomo di carbonio legato a un altro atomo di carbonio e a tre atomi di idrogeno
Carbonio secondario: In una molecola, un atomo di carbonio legato a due atomi di carbonio e a due atomi di idrogeno
Carbonio quaternario: In una molecola, un atomo di carbonio legato a quattro atomi di carbonio
Carbonio terziario: In una molecola, un atomo di carbonio legato a tre atomi di carbonio e a un atomo di idrogeno
Carbossile: Gruppo funzionale organico avente la struttura ‑COOH
Catalizzatore: Sostanza che accelera la velocità di una reazione chimica e che può essere recuperata praticamente inalterata alla fine della reazione
Catalizzatore di Lindlar: Catalizzatore per l’idrogenazione catalitica di un alchino ad alchene cis. Il catalizzatore di Lindlar è Pd assorbito su CaCO3 contenente acetato di piombo (II) e chinolina
Catione: Ione carico positivamente che ha origine da un atomo neutro quando esso perde uno o più elettroni
Centro chirale: Atomo di carbonio al quale sono legati quattro gruppi diversi
b-Chetoestere: Composto organico contenente un carbonile chetonico sul carbonio b rispetto al carbonile dell’estere
Chetone: Composto organico avente un carbonile con due gruppi alchilici e/o arilici legati al carbonio carbonilico
Chimica Organica: Branca della chimica che studia i composti del carbonio
Chirale: Avente la proprietà di non essere sovrapponibile alla propria immagine speculare
Cianidrina: Gruppo funzionale avente un gruppo ossidrile ‑OH e un gruppo ciano ‑CN sullo stesso atomo di carbonio. Si ottiene dall’addizione di HCN al carbonile di un’aldeide o un chetone
Cianuro: Anione nucleofilo avente la struttura -C≡N
Ciclo-: Prefisso utilizzato nella nomenclatura IUPAC per indicare una struttura ciclica
Cicloalcano: Idrocarburo i cui gli atomi di carbonio formano uno o più anelli.
Cicloesano a barca: Conformazione del cicloesano rassomigliante a una barca. Il cicloesano a barca non ha tensione angolare, ma possiede un gran numero di interazioni eclissanti che lo rendono meno stabile del cicloesano a sedia
Cicloesano a sedia: Conformazione del cicloesano che rassomiglia a una sedia. è la conformazione a più bassa energia della molecola
Cinetica chimica: Ramo della chimica che studia la velocità di reazione, in particolare il rapporto tra velocità e concentrazione
Cinetica del primo ordine: Si ha quando la velocità della reazione dipende dalla concentrazione di uno solo dei reagenti
Cinetica del secondo ordine: Si ha quando la velocità della reazione dipende dalla concentrazione di due reagenti
Cis: Prefisso che indica che in un anello o in un doppio legame carbonio-carbonio bisostituiti i due sostituenti sono dalla stessa parte
Cloridrina: Composto avente un atomo di cloro e un gruppo ossidrilico su atomi di carbonio adiacenti
Clorurazione: Reazione di un composto organico con Cl2
Cloruro acilico: Composto caratterizzato dal gruppo funzionale -COCl
Combustione: Reazione di ossido-riduzione nella quale un alcano o un altro composto organico reagisce con l’ossigeno per formare anidride carbonica, acqua e liberando energia sotto forma di calore
Composto alifatico: Termine usato per indicare idrocarburi non aromatici (alcani, alcheni ed alchini) ed i loro derivati
Composto aromatico policiclico: Composto con due o più anelli aromatici benzenoidi fusi assieme
Composto carbonilico a-b insaturo: Composto contenente un doppio legame C=C e un doppio legame C=O separati da un solo legame semplice, quindi coniugati
Composto b-idrossicarbonilico: Composto organico caratterizzato da un gruppo ossidrile sul carbonio in b rispetto al gruppo carbonilico
Composto insaturo: Composto con più di un legame tra atomi adiacenti, solitamente atomi di carbonio
Composto meso: Stereoisomero otticamente inattivo per compensazione interna (due sostituenti del centro chirale sono l’uno l’immagine speculare dell’altro e non sono sovrapponibili). Nella molecola esiste un piano di simmetria
Composto organometallico: Composto nel quale un atomo di carbonio è legato a un atomo di metallo
Composto otticamente attivo: Composto in grado di far ruotare il piano della luce polarizzata qualora questa attraversi una soluzione del composto
Composto otticamente inattivo: Composto non in grado di far ruotare il piano della luce polarizzata qualora questa attraversi una soluzione del composto
Condensazione aldolica: Reazione in cui due molecole di aldeide o chetone reagiscono l’una con l’altra in presenza di una base per formare un composto b-idrossicarbonilico
Condensazione aldolica incrociata: Formazione di un composto b-idrossicarbonilico (b-aldolo) per condensazione, in ambiente alcalino, di un aldeide o di un chetone aventi idrogeni in a con un’altra aldeide o un altro chetone privi di idrogeni in a
Condensazione di Claisen: Reazione in cui due molecole di un estere reagiscono in presenza di una base per formare un b-chetoestere
Condensazione di Claisen incrociata: Reazione di Claisen in cui i due esteri reagenti sono diversi
Configurazione: Disposizione tridimensionale di atomi che caratterizza un determinato stereoisomero
Configurazione assoluta: Esatta struttura tridimensionale di una molecola chirale. Essa viene specificata verbalmente dalla convenzione R,S di cahn-Ingold-Prelog
Conformazione a barca: Conformazione instabile adottata dal cicloesano simile a una barca. La sua instabilità è dovuta alla tensione torsionale e alla tensione sterica
Conformazione a sedia: Conformazione stabile adottata dal cicloesano che ricorda una sedia. La sua stabilità è dovuta alla completa eliminazione della tensione angolare (tutti gli angoli C-C-C sono di 109,5°) e tensione torsionale (tutti i gruppi situati su carboni adiacenti sono sfalsati l’uno rispetto all’altro)
Conformazione eclissata: Conformazione di una molecola in cui i legami su un atomo di carbonio sono perfettamente allineati con quelli dell’atomo di carbonio adiacente
Conformazione sfalsata: Disposizione particolare degli atomi dovuta a rotazione intorno al legame semplice C-C tale che, osservando lungo l’asse del legame C-C, ciascun gruppo o atomo legato a uno dei due carboni sta al centro del “vuoto” tra due gruppi o atomi legati all’altro carbonio
Conformazioni: Differenti disposizioni degli atomi di una molecola che interconvertono mediante rotazione intorno a legami singoli C-C
Coniugazione: Sistema di atomi uniti da legami covalenti con legami singoli e multipli che si alternano.
Convenzione R,S: Metodo per la definizione della configurazione assoluta di un centro chirale che si basa sull’applicazione delle regole di Cahn-Ingold-Prelog
Coppia di elettroni non di legame: Coppia di elettroni di valenza che non è coinvolta nella formazione di un legame covalente (si parla anche di coppie solitarie o doppietti non condivisi)
Coppia ionica: In un solvente, coppia di ioni con segno opposto in cui nessuna molecola di solvente separa il catione dall’anione. È detta anche: coppia ionica intima
Costante di acidità: Valore che rappresenta la forza di un acido (Ka)
Costante di equilibrio: Espressione matematica che correla le quantità di reagenti e prodotti presenti all’equilibrio (Ke)
Costante di velocità: La costante k in un’equazione cinetica
Decarbossilazione: Reazione che comporta perdita di CO2 da una molecola
Deidroalogenazione: reazione di eliminazione in cui gli elementi idrogeno e alogeno sono persi da un materiale di partenza
Delocalizzazione : Situazione che si riscontra in un sistema elettronico in cui gli elettroni di legame non sono localizzati tra due atomi come in un legame semplice, ma esiste una dispersione della densità elettronica su un sistema p coniugato
Derivati funzionali degli acidi carbossilici: Composti che formalmente derivano dagli acidi carbossilici per sostituzione del gruppo ossidrile con un atomo o un gruppo di atomi
Destrorotatorio: Termine usato per descrivere una sostanza otticamente attiva che ruota il piano di polarizzazione della luce polarizzata nella direzione destrorsa (senso orario). Si dice anche: destrogiro
Diagramma di energia: Rappresentazione schematica delle variazioni di energia che hanno luogo quando i reagenti si convertono nei prodotti. Un diagramma di energia indica quanto facilmente procede una reazione, quanti stadi sono coinvolti e quale è l’energia relativa di reagenti, prodotti e intermedi
Dialogenuro vicinale: composto che ha due atomi di alogeno legati ad atomi di carbonio adiacenti
Diastereoisomeri: Stereoisomeri che non sono l’uno l’immagine speculare dell’altro
Diazotazione: Trasformazione di un’ammina primaria, RNH2, in uno ione diazonio, RN2+, per trattamento con acido nitroso
Diene: Idrocarburo contenente due doppi legami
Diene coniugato: Composto con due doppi legami C=C separati da un legame semplice C-C
Diene cumulato: Diene in cui due doppi legami condividono uno stesso atomo di carbonio (C=C=C)
Diene isolato: Composto contenente due doppi legami C=C separati da più di un legame semplice C-C
Diolo: Composto organico caratterizzato da due gruppi ossidrilici
Dipolo: Separazione di carica elettrica
Disidratazione: Reazione che causa la perdita degli elementi dell’acqua dai reagenti
Disolfuro: Composto contenente un legame S-S
E2: Eliminazione bimolecolare, meccanismo di eliminazione che procede secondo un processo a stadio unico concertato, in cui entrambi i reagenti sono coinvolti nello stato di transizione.
E1: Eliminazione monomolecolare, meccanismo di eliminazione che procede a due stadi, con un carbocatione intermedio. Nel passaggio che determina la velocità del processo solo una specie subisce variazioni di covalenza
Effetti sterici: Effetti destabilizzanti che derivano dall’impossibilità di due gruppi o atomi di occupare la stessa zona dello spazio
Effetto induttivo: Spostamento di densità elettronica attraverso i legami s causata dalla differenza di elettronegatività degli atomi
Effetto induttivo di attrazione elettronica: Effetto induttivo in cui un atomo elettronegativo attira verso di sé densità elettronica attraverso i legami s
Effetto induttivo di elettron donatore: Effetto induttivo in cui un atomo elettropositivo o un gruppo polarizzabile dona densità elettronica ad un altro atomo attraverso i legami s
Effetto orientante: Capacità di un sostituente del benzene di indirizzare un’ulteriore sostituzione in orto, para o meta
Elettrofilo: Ogni atomo, molecola o ione che accetta una coppia di elettroni da un nucleofilo per formare con esso un nuovo legame covalente
Elettronegatività: Misura della forza di attrazione di un atomo nei confronti degli elettroni che condivide con un altro atomo attraverso un legame chimico
Elettroni di valenza: Gli elettroni che si trovano nel “livello energetico” più esterno
Eliminazione: Reazione chimica in cui una molecola si scinde in due nuove molecole attraverso la rottura di due legami s e la formazione di un legame p
b-Eliminazione: Reazione di eliminazione in cui avviene la rimozione di atomi o gruppi di atomi da carboni adiacenti per formare un doppio o triplo legame
Emiacetale: Composto contenente un gruppo alcossido –OR e un gruppo ossidrilico –OH legati allo stesso atomo di carbonio
Enammina: Composto organico avente un azoto amminico legato a un doppio legame C=C (R2N-CR=CR2)
Enantiomeri: Stereoisomeri che sono immagini speculari non sovrapponibili
Endoergonico: Reazione con variazione positiva dell’energia libera, che pertanto non è spontanea
Endotermica: Reazione che assorbe calore e pertanto ha una variazione positiva dell’entalpia
Energia di attivazione: Differenza di energia potenziale tra i reagenti e lo stato di transizione attraverso il quale essi vanno a prodotti. Determina la velocità con cui la reazione procede.
Energia di dissociazione di legame: Quantità di energia necessaria per scindere omoliticamente un legame covalente
Energia di risonanza: La differenza di energia tra un ibrido di risonanza e la struttura limite più stabile che contribuisce alla risonanza in cui la densità elettronica è localizzata su particolari atomi o legami covalenti
Energia di torsione: Energia richiesta per ruotare le molecole intorno al legame tra due atomi
Enolato: Anione stabilizzato per risonanza, formato quando una base rimuove un idrogeno da un carbonio in a a un carbonile
Enolo: Composto organico in cui un gruppo OH è legato ad un carbonio impegnato in doppio legame C=C
Entgegen (E): Termine usato per descrivere la stereochimica di un doppio legame C=C in cui i gruppi ad alta priorità su ciascun carbonio si trovano su lati opposti del doppio legame
Entropia: Misura del disordine di un sistema. Una definizione più corretta è la seguente: Funzione dello stato di un sistema termodinamico la cui variazione in un processo reversibile infinitesimale è uguale al rapporto tra il calore che il sistema assorbe dall’ambiente circostante e la temperatura assoluta
Epossido: Etere ciclico avente l’atomo di ossigeno inserito in un anello triatomico. È detto anche ossirano
Equazione cinetica: Equazione che esprime la dipendenza della velocità di una reazione dalla concentrazione dei reagenti
Equazione di velocità del primo ordine: Equazione di velocità in cui compare la concentrazione di uno solo dei reagenti
Equazione di velocità del secondo ordine: Equazione in cui la velocità della reazione è direttamente proporzionale alla concentrazione di due reagenti
Esoergonica: Reazione con variazione negativa dell’energia libera, che pertanto è spontanea
Esotermica: Reazione che rilascia calore e pertanto ha una variazione negativa dell’entalpia
Estere: Classe di composti organici contenente il gruppo funzionale -COOR
Esterificazione: Una reazione che converte un acido carbossilico o un derivato dell’acido carbossilico in un estere
Esterificazione di Fischer: Una reazione, catalizzata da un acido forte, che converte un acido carbossilico e un alcool per dare un estere
Etere: Gruppo funzionale che possiede due gruppi organici legati allo stesso atomo di ossigeno
Etere non simmetrico: Etere nel quale i due gruppi organici legati all’ossigeno sono differenti
Etere simmetrico: Etere nel quale i due gruppi organici legati all’ossigeno sono uguali
Eteroatomo: Atomo diverso dal carbonio e dall’idrogeno in una molecola organica
Eterociclo: Composto i cui anelli sono formati, oltre che da atomi di carbonio, da uno o più atomi diversi
Eterociclico aromatico: Composto organico ciclico con uno o più atomi diversi dal carbonio come costituenti dell’anello che, pur possedendo legami multipli tra gli atomi dell’anello, subisce più facilmente reazioni di sostituzione che di addizione
Eterolisi: Rottura di un legame chimico covalente (a due elettroni) in cui entrambi gli elettroni rimangono su uno solo degli atomi. È detta anche scissione eterolitica
Etinile: Sostituente alchinilico avente la struttura -C≡C-H
E, Z:Sistema di nomenclatura utilizzata per indicare in maniera non ambigua gli stereoisomeri degli alcheni
FANS: Farmaco antinfiammatorio non steroideo come l’aspirina o l’ibuprofene
Fenolo: Composto organico che contiene un gruppo ossidrile legato a un anello aromatico
Feromone: Sostanza chimica utilizzata nelle specie animali e anche vegetali per trasmettere dei messaggi
Forma di risonanza: Struttura di Lewis individuale di un ibrido di risonanza
Formazione di legame eterogenico: Ciò che succede quando solo uno dei reagenti dona entrambi gli elettroni nel formare un nuovo legame
Formazione di legame omogenico: Ciò che succede quando ciascun reagente dona un elettrone nel formare un nuovo legame
Formile: Gruppo funzionale avente la struttura H-C=O
Formula bruta: Formula chimica di un composto in cui sono indicati il tipo e il numero degli atomi che lo costituiscono ma non la sua struttura
Formula chimica: Sistema stenografico utile a porre in evidenza, rapidamente, quali elementi fanno parte si una molecola e in quale rapporto di combinazione
Formula di struttura: Formula chimica che indica la disposizione spaziale degli atomi in una molecola, mostrando come gli atomi sono legati tra loro e con quale tipo di legame
Formule di proiezione di Fischer: Convenzione per rappresentare la configurazione di uno stereocentro in due dimensioni. Il centro dello stereocentro è rappresentato all’intersezione di un segmento verticale e di uno orizzontale. I legami verticali allo stereocentro sono quelli che nella struttura tridimensionale si allontanano dall’osservatore, mentre quelli orizzontali sono diretti verso l’osservatore
Formule limite di risonanza: Nella teoria della risonanza, due o più strutture che differiscono solamente per la distribuzione degli elettroni
Forza di legame: Termine alternativo a energia di dissociazione di legame
Forze di Van der Waals: Forze intermolecolari molto deboli responsabili dell’associazione tra le molecole non polari o poco polari allo stato liquido e solido
Forze intermolecolari: Tipi di interazioni di natura elettrostatica che si instaurano tra le molecole
Freccia ad amo o a semipunta: Freccia curva a semipunta usata per indicare il movimento di un singolo elettrone nella descrizione di un meccanismo di reazione
Freccia curva a punta intera: Freccia curva utilizzata in un meccanismo di reazione per indicare il movimento di una coppia elettronica
Glicol: Composto con due gruppi ossidrilici su atomi di carbonio adiacenti
Grado di insaturazione: Numero di anelli e/o di legami p contenuti in una molecola. Il grado di insaturazione confronta il numero di idrogeni in un composto con quello in un idrocarburo saturo contenente lo stesso numero di atomi di carbonio
Gruppo acetile: Sostituente avente la struttura –COCH3
Gruppo acile: Sostituente che ha la struttura generale -COR
Gruppo alchilico: Gruppo organico ottenuto formalmente per rimozione di un atomo di idrogeno da un idrocarburo saturo aciclico, può essere rappresentato con il simbolo R-
Gruppo allile: Sostituente avente la struttura ‑CH2CH=CH2
Gruppo alcossile: Sostituente contenente un gruppo alchilico legato a un ossigeno (-OR)
Gruppo attivante: Gruppo elettrondonatore che, quando è legato a un anello aromatico, ne aumenta la reattività verso la sostituzione elettrofila aromatica
Gruppo benzile: Sostituente avente la struttura C6H5CH2-
Gruppo benzoile: Sostituente avente la struttura C6H5CO-
Gruppo carbonilico: Il gruppo funzionale C=O
Gruppo ciano: Il gruppo funzionale -C≡N
Gruppo disattivante: Gruppo elettronattrattore che, quando è legato a un anello aromatico, ne riduce la reattività verso la sostituzione elettrofila aromatica
gruppo elettronattrattore: Atomo o gruppo di atomi legato con legame covalente al resto della molecola avente la capacità di attrarre parzialmente verso di sé la nuvola elettronica associata al legame
Gruppo fenile: Termine usato per indicare il gruppo ‑C6H5 quando l’anello benzenico viene considerato come sostituente
Gruppo formile: Il gruppo funzionale -CHO
Gruppo funzionale: Atomo o gruppo di atomi, parte di una molecola più grande, con peculiari proprietà fisiche e chimiche e dalla reattività chimica caratteristica
Gruppo mercapto: Termine alternativo per il gruppo tiolico ‑SH
Gruppo meta orientante: Gruppo legato ad un anello benzenico che, nel corso di una sostituzione elettrofila aromatica, dirige preferenzialmente il gruppo entrante nella posizione meta
Gruppo metilenico: Gruppo CH2 legato a una catena di carboni (-CH2-) o facente parte di un doppio legame (CH2=)
Gruppo orto-para orientante: Gruppo legato ad un anello benzenico che nel corso di una sostituzione elettrofila aromatica dirige preferenzialmente il gruppo entrante nelle posizioni orto e para
Gruppo R: Abbreviazione generalizzata per una struttura organica parziale
Gruppo uscente: Atomo o gruppo di atomi con carica o senza che si separano da un atomo di carbonio nel corso di una reazione di sostituzione o di eliminazione e in grado di accettare il doppietto elettronico del legame
Gruppo vinilico: Sostituente alchenilico che ha la struttura ‑CH=CH2
Ibridazione: Combinazione matematica di due o più orbitali atomici (aventi forme differenti) per formare un ugual numero di orbitali ibridi (tutti con la medesima forma)
Ibrido di risonanza: Molecola che non può essere adeguatamente rappresentata da un’unica struttura, ma deve invece essere considerata come una media di due o più strutture di risonanza. Le strutture di risonanza differiscono tra loro solo per la posizione degli elettroni p e dei doppietti elettronici solitari, non dei nuclei.
Idratazione: Addizione di acqua a una molecola
Idroalogenazione: Addizione elettrofila di acido alogenidrico a un alchene o a un alchino
Idrocarburi aromatici: Classe di idrocarburi ciclici caratterizzati da forte insaturazione e alta energia di risonanza in cui prevalgono le reazioni di sostituzione rispetto a quelle di addizione
Idrocarburi aromatici ad anelli condensati: Idrocarburi formati da più anelli benzenici che hanno in comune due o più atomi di carbonio
Idrocarburo: Composto organico formato solo da atomi di carbonio e di idrogeno
Idrocarburo insaturo: Idrocarburo che contiene un numero di atomi di idrogeno minore rispetto al numero massimo di atomi di idrogeno per atomo di carbonio presente
Idrocarburo saturo: Composto che contiene solo legami di tipo s C-C e C-H, nessun anello e avente quindi il massimo numero di atomi di idrogeno legati ad atomi di carbonio
Idrofilo: Attratto dall’acqua
Idrofobo: Non attratto dall’acqua
Idrogenazione: Addizione di idrogeno a un doppio o triplo legame per dare un prodotto saturo
Idrogenazione catalitica: Reazione di riduzione che comporta l’addizione di una molecola di H2 a un legame p in presenza di un catalizzatore metallico
a-Idrogeni: Atomi di idrogeno legati al carbonio adiacente a quello del gruppo funzionale
Idrogeno primario: Atomo di idrogeno che è legato a un atomo di carbonio primario
Idrogeno secondario: Atomo di idrogeno che è legato a un atomo di carbonio secondario
Idrogeno terziario: Atomo di idrogeno che è legato a un atomo di carbonio terziario
Idrolisi: Reazione di scissione di una molecola che ha origine dall’attacco dell’acqua
Idruro: Ione idrogeno carico negativamente (H:-)
Idruro metallico: Reagente che contiene un legame polarizzato tra idrogeno e un metallo, tale da indurre una carica negativa parziale sull’atomo di idrogeno
Immagine speculare: Il riflesso di un oggetto in uno specchio
Immina: Composto, detto anche Base di Schiff, caratterizzato dal gruppo funzionale C=N
Ingombro sterico: La capacità dei gruppi, legata alle loro dimensioni, di ostacolare o impedire l’accesso ad un sito di reazione in una molecola
Inibitore di radicali: Composto che impedisce alle reazioni radicaliche di avvenire
Interazione 1,3-diassiale: Energia di tensione provocata da interazione sterica tra atomi o gruppi uniti ai tre legami assiali posti sullo stesso lato della molecola nel cicloesano a sedia
Interazione dipolo-dipolo: Forza attrattiva intermolecolare tra due dipoli permanenti di molecole polari. L’interazione si instaura tra un estremo positivo di un dipolo e uno negativo di un altro dipolo
Intermedio: Specie che di forma durante una reazione a più stadi, ma che non è il prodotto finale. Gli intermedi sono più stabili degli stati di transizione, ma non sono abbastanza stabili da poter essere isolati
Inversione della sedia: Processo a due stadi in cui una conformazione a sedia del cicloesano si converte in una conformazione opposta, anch’essa a sedia
Inversione di configurazione: Configurazione stereochimica opposta rispetto a un centro stereogenico nel reagente e nel prodotto di una reazione chimica
Ione acetato: Ione derivante dalla dissociazione dell’acido acetico: CH3COO-
Ione Acilio: Elettrofilo carico positivamente,stabilizzato per risonanza, R-C+=O ↔ R-C≡O+, formato quando l’acido di lewis AlCl3 ionizza il legame carbonio-alogeno di un cloruro acilico
Ione alcossido: Ione RO- ottenuto per deprotonazione di un alcool ad opera di metalli o idruri metallici
Ione alonio: Specie contenente un alogeno carico positivamente. Uno ione alonio a ponte contiene un anello triatomico e si forma nell’addizione di alogeno a un alchene
Ione ammonico: Composto con carica positiva in cui allo stesso atomo di azoto sono legati quattro atomi di idrogeno.
Ione alchil o arilammonico: Composto con carica positiva in cui allo stesso atomo di azoto sono legati quattro gruppi di cui almeno uno è un atomo di idrogeno e fino a tre sono gruppi alchilici e/o arilici
Ione ammonico quaternario: Composto con carica positiva in cui allo stesso atomo di azoto sono legati quattro gruppi alchilici e/o arilici
Ione enolato: Anione de localizzato derivato dalla rimozione di un protone da un enolo o dal composto carbonilico in equilibrio con l’enolo.
Ione nitronio: Elettrofilo avente la struttura NO2+, coinvolto nel processo di nitrazione dei composti aromatici, che si forma attraverso un equilibrio acido-base tra acido solforico (l’acido) e acido nitrico (la base): HONO2 + 2H2SO4® H3O+ + 2HSO4- + +NO2
Ione nitrosonio: Ione di formula NO+
Ione tiolato: Ione RS- ottenuto per deprotonazione di un tiolo ad opera di basi
Isomeria: Proprietà di certi composti chimici che, pur essendo costituiti da un ugual numero e tipo di atomi, hanno strutture e proprietà fisiche e chimiche differenti
Isomeri di struttura o costituzionali: Composti diversi aventi uguale formula bruta ma differente formula di struttura
Isomeri geometrici: Stereoisomeri che differiscono tra di loro per la posizione dei sostituenti sia in una struttura ciclica che in un doppio legame
Isomero cis: In un anello o in un doppio legame bisostituito, isomero che mostra i due sostituenti dalla stessa parte dell’anello o del doppio legame
Isomero meta: Anello benzenico bisostituito nel quale i due sostituenti si trovano separati da un atomo di carbonio dell’anello. Si indica anche come benzene 1,3-disostituito
Isomero orto: Anello benzenico bisostituito nel quale i due sostituenti si trovano su due atomi di carbonio adiacenti dell’anello. Si indica anche come benzene 1,2-disostituito
Isomeri ottici: Termine alternativo per enantiomeri e diasteroisomeri
Isomero para: Anello benzenico bisostituito nel quale i due sostituenti si trovano separati da altri due atomi di carbonio dell’anello. Si indica anche come benzene 1,4-disostituito
Isomero trans: In un anello o in un doppio legame bisostituito, isomero che mostra i due sostituenti dalla parte opposta dell’anello o del doppio legame
Isotopi: Atomi dello stesso elemento con diverso numero di massa
IUPAC: International Union of Pure and Applied Chemistry (in italiano Unione Internazionale di Chimica Pura ed Applicata)
Legame: Unione di due atomi in un arrangiamento elettronico stabile. La formazione di un legame è un processo favorevole che porta a una diminuzione dell’energia e a un aumento di stabilità
Legame covalente: Legame risultante dalla condivisione di elettroni tra due nuclei. Ogni legame covalente è a due elettroni
Legame covalente polare: Legame covalente in cui la distribuzione elettronica tra gli atomi è asimmetrica a causa della loro differente elettronegatività
Legame idrogeno: Attrazione elettrostatica tra un atomo di idrogeno legato con legame covalente a un atomo molto elettronegativo (F, O o N) e il doppietto solitario di un atomo molto elettronegativo (F, O o N) di una molecola adiacente (legame a idrogeno intermolecolare) o della stessa molecola (legame a idrogeno intramolecolare)
Legame ionico: Legame ottenuto dal trasferimento di elettroni da un elemento a un altro. Il legame ionico è il risultato della forte attrazione elettrostatica tra gli ioni di carica opposta
Legame non polare: Legame covalente nel quale gli elettroni sono distribuiti in maniera uguale tra i due atomi
Legame polare: Legame covalente nel quale gli elettroni non sono distribuiti in maniera uguale tra i due atomi a causa della maggiore elettronegatività di uno di essi
Legame s: Legame covalente formato per sovrapposizione testa a testa di orbitali atomici. è detto anche legame semplice
Legame p: Legame formato dalla sovrapposizione lato-su-lato di due orbitali p, nel quale la densità elettronica non è localizzata sull’asse di congiungimento dei due nuclei
Legami assiali del cicloesano: Nella conformazione a sedia del cicloesano si definiscono assiali i sei legami degli atomi di idrogeno giacenti sopra o sotto il piano medio degli atomi di carbonio dell’anello, cioè sopra e sotto l’equatore dell’anello
Legami equatoriali del cicloesano: Nella conformazione a sedia del cicloesano si definiscono equatoriali i sei legami degli atomi di idrogeno giacenti più vicini al piano medio degli atomi di carbonio dell’anello, cioè che si trovano in una zona intorno all’equatore dell’anello
Levorotatorio: Termine per indicare che un composto, posto in un polarimetro, ruota verso sinistra il piano della luce polarizzata, ovvero in direzione antioraria
Luce polarizzata nel piano: Luce ordinaria le cui onde elettromagnetiche oscillano in un singolo piano invece che in piani casuali. Si può ottenere da un fascio di luce ordinaria attraverso un filtro polarizzatore
Lunghezza di legame: Distanza media tra i nuclei di due atomi legati
Mappa di potenziale elettrostatico: Mappatura convenzionale a colori che illustra la distribuzione della densità elettronica di una molecola. Le regioni elettron-ricche sono indicate in rosso, mentre quelle elettron-povere in blu. Le regioni con densità elettronica intermedie sono indicate in arancione, giallo e verde
Meccanismo a catena: Meccanismo di reazione che implica la ripetizione dei singoli stadi
Meccanismo di reazione: Descrizione dettagliata di come i legami si rompono e si formano mentre i reagenti si convertono nei prodotti
Meccanismo SN1: Sostituzione nucleofila monomolecolare; monomolecolare significa che nel passaggio determinante è coinvolta solo una molecola. Meccanismo di sostituzione nucleofila che procede in un processo a due stadi che coinvolge un intermedio di tipo carbocationico
Meccanismo SN2: Sostituzione nucleofila bimolecolare. Meccanismo di sostituzione nucleofila che avviene per un processo concertato, dove entrambi i reagenti sono coinvolti nello stato di transizione
Meta orientante: Sostituente presente sull’anello aromatico in grado di orientare in posizione meta l’entrata di un elettrofilo nella reazione di sostituzione elettrofila aromatica
Metilazione: Reazione nella quale un gruppo -CH3 viene trasferito da un composto a un altro
Miscela racemica: Miscela equimolecolare di due enantiomeri, otticamente inattiva
Molecola: Insieme neutro di atomi tenuti insieme da legami covalenti
Molecola apolare: Molecola che non possiede un dipolo netto: una molecola apolare non possiede legami polari, o possiede legami polari i cui dipoli opposti si annullano
Molecola chirale: Molecola non sovrapponibile alla propria immagine speculare
Molecola polare: Molecola che ha un dipolo netto. Una molecola polare possiede un legame polare o legami polari multipli i cui dipoli si rafforzano
Momento dipolare (m): Misura della polarità di una molecola. Si ha momento dipolare quando i centri di massa delle cariche positive e negative in una molecola non coincidono
Monoalogenazione: Reazione di alogenazione che coinvolge la sostituzione di un solo atomo di idrogeno con un atomo di alogeno
Nitrazione: Reazione di sostituzione elettrofila aromatica in cui un derivato benzenico reagisce con uno ione nitronio per dare il nitrobenzene derivato
Nitrile: Composto caratterizzato dal gruppo funzionale ‑C≡N
N-Nitrosoammina: Composto organico che ha la struttura generale R2N-N=O. Si ottiene per reazione di un’ammina secondaria con lo ione nitronio
Nome comune: Nome di una molecola adottato prima del sistema di nomenclatura iUPAC
Nome sistematico: Nome chimico di una molecola in grado di indicarne la sua struttura. Viene detto anche nome IUPAC
Normal alcano: Alcano aciclico costituito da atomi di carbonio legati tutti a un’unica catena lineare
Nucleofilo: Reagente basico, ricco di elettroni, che tende ad attaccare il nucleo del carbonio; dona infatti una coppia di elettroni ad un elettrofilo in una reazione polare di formazione di legame
Numero atomico: Il numero di protoni nel nucleo di un atomo
Numero di massa: Il numero totale di protoni e neutroni presenti nel nucleo di un particolare atomo
“Ogni simile scioglie il suo simile”: Principio secondo il quale i composti si sciolgono in solventi che mostrano interazioni non di legame dello stesso tipo: cioè composti polari si sciolgono in solventi polari, composti non polari si sciolgono in solventi non polari
Olefina: Alchene. Composto organico che possiede il gruppo funzionale doppio legame
Omolisi: Rottura di un legame covalente che suddivide equamente gli elettroni tra i due atomi del vecchio legame. L’omolisi genera radicali intermedi privi di carica
Orbitali ibridi sp: Orbitali atomici formati dalla combinazione matematica di un orbitale s e un orbitale p
Orbitali ibridi sp2: Orbitali atomici formati dalla combinazione matematica di un orbitale s e due orbitali p
Orbitali ibridi sp3: Orbitali atomici formati dalla combinazione matematica di un orbitale s e tre orbitali p
Ordine della velocità di una reazione: Somma degli esponenti dei termini di concentrazione presenti nell’equazione cinetica di una reazione
Orto orientante: Sostituente di un anello benzenico che orienta l’entrata di un nuovo gruppo in posizione orto nella reazione di sostituzione elettrofila aromatica
Ossidazione: Processo in cui un composto perde elettroni. Per un composto organico, l’ossidazione porta all’aumento di legami C-O o alla riduzione dei legami C-H presenti
Ossidrilazione: Addizione di due gruppi ossidrilici (-OH) a un doppio legame
Ossidrile: Gruppo funzionale -OH
Ossima: Composto con gruppo funzionale C=NOH che si ottiene per reazione tra un aldeide o un chetone e l’idrossilammina
Ossirano: Etere ciclico a tre termini in cui l’atomo di ossigeno è parte dell’anello. è detto anche epossido
Para orientante: Sostituente di un anello benzenico che orienta l’entrata di un nuovo gruppo in posizione para nella reazione di sostituzione elettrofila aromatica
Passaggio determinante di una reazione multistadio: Passaggio notevolmente più lento che regola la velocità complessiva della reazione
Peracido o perossiacido: Composto con il gruppo funzionale –CO3H
Perossido: Molecola contenente un legame ossigeno-ossigeno come gruppo funzionale (RO‑OR)
Petrolio: Combustibile fossile contenente una miscela complessa di composti, principalmente idrocarburi che contengono da uno a 40 atomi di carbonio
Piano di simmetria: Piano che taglia a metà un oggetto (o una molecola) in modo tale che una delle parti risultanti sia l’immagine speculare dell’altra metà
pKa: Scala logaritmica della forza degli acidi. pKa = -logKa. Minore è il pKa più forte è l’acido
Polarimetro: Strumento che misura il valore della rotazione della luce piano-polarizzata determinata da un composto organico otticamente attivo
Polarità: Distribuzione asimmetrica di elettroni in una molecola che si ha quando un atomo attrae elettroni più fortemente di un altro
Polarizzabilità: Misura della variazione della distribuzione elettronica in una molecola in risposta a cambiamenti nelle interazioni elettriche con solventi o reagenti ionici o polari
Policiclico: Composto contenente più di un anello
Poliene: Composto organico che contiene tre o più doppi legami
Potere rotatorio specifico: Per una sostanza otticamente attiva, a una data temperatura e una data lunghezza d’onda, l’angolo di rotazione del piano di vibrazione della luce polarizzata quando il cammino ottico e la concentrazione sono unitari
Primario, secondario, terziario: Termini usati per descrivere il tipo di sostituzione ad un certo sito. Un sito primario ha solo un sostituente organico legato ad esso, un sito secondario ne ha due, un sito terziario ne ha tre ed un sito quaternario ne ha quattro
Prodotto di addizione 1,2: Prodotto risultante dall’addizione di due gruppi su due atomi adiacenti di un sistema coniugato
Prodotto di addizione 1,4: Prodotto risultante dall’addizione di due gruppi agli atomi in posizione 1 e 4 di un sistema coniugato
Prodotto naturale: Composto organico isolato da una fonte naturale
Propagazione: Stadio di una reazione radicalica a catena nel quale un radicale reagisce con un reagente per formare un nuovo radicale e il prodotto della reazione. La propagazione continua sino a quando avvengono gli stadi di terminazione del processo radicalico
Protone: Atomo di idrogeno che possiede una carica positiva
Racemizzazione: Processo mediante il quale una forma otticamente attiva di una sostanza viene convertita in una miscela otticamente inattiva costituita da quantità uguali di isomeri destrogiro e levogiro dello stesso composto iniziale o di uno differente
Racemo: Miscela, otticamente inattiva, composta da parti uguali dei due enantiomeri di una sostanza chirale
Radicale libero: atomo (o gruppo di atomi) con un elettrone spaiato (o dispari) che costituisce un intermedio reattivo e si ottiene per omolisi di un legame covalente
Reagente: Sostanza chimica con la quale un composto organico reagisce
Reagente di Feeling: Reagente che ossida le aldeidi ad acidi carbossilici utilizzando un sale di Cu2+ come ossidante. Come prodotto collaterale di forma un precipitato rosso mattone di Cu2O
Reagente organometallico: Reagente che contiene un atomo di carbonio legato a un metallo
Reagente di Tollens: Reagente che ossida aldeidi ad acidi carbossilici, a base di ossido di argento(I) sciolto in una soluzione di idrossido di ammonio. Il test è positivo se si deposita uno specchio di argento sulle pareti del recipiente di reazione
Reattivo di Grignard: Alogenuro di organomagnesio; reagente organometallico di struttura R-Mg-X,
Reazione a catena: Una reazione che, una volta iniziata, continua ripetendo una serie di stadi di propagazione di catena. Gli stadi di propagazione di catena sono quelli in cui si formano i prodotti di una reazione a catena
Reazione acido-base di Lewis:. Reazione che avviene quando una base di Lewis dona una coppia elettronica a un acido di Lewis formando con esso un legame covalente
Reazione bimolecolare: Reazione in cui la concentrazione di entrambi i reagenti influisce sulla velocità di reazione ed entrambi compaiono nell’equazione di velocità. In una reazione bimolecolare, due reagenti sono coinvolti nell’unico stadio cineticamente determinante
Reazione concertata: Reazione in cui la formazione e la scissione dei legami avviene in un unico stadio
Reazione del primo ordine: Reazione la cui velocità dipende dalla concentrazione di un solo reagente
Reazione del secondo ordine: Reazione la cui velocità dipende dalla concentrazione di due reagenti
Reazione di addizione: Reazione che avviene quando due reagenti si sommano per formare un solo nuovo prodotto, senza atomi “che avanzano”
Reazione di addizione elettrofila: Addizione di un elettrofilo ad un alchene a dare un prodotto saturo
Reazione di addizione nucleofila al carbonile: Reazione in cui un nucleofilo si somma al gruppo carbonilico elettrofilo di un chetone o di un’aldeide a dare un alcool
Reazione di disidratazione: Reazione in cui un singolo reagente si divide in due prodotti
Reazione di eliminazione: Ciò che avviene quando un singolo reagente di scinde in due prodotti, uno con grado di insaturazione superiore
Reazione di b-eliminazione: Rimozione di atomi o gruppi di atomi da due atomi di carbonio adiacenti per formare un doppio legame C=C
Reazione di Friedel-Crafts: Sostituzione elettrofila aromatica per alchilare o acilare un anello aromatico
Reazione di Sandmeyer: Reazione di sostituzione nucleofila di un sale di arildiazonio con un alogenuro rameoso a dare un alogenuro arilico
Reazione di sostituzione: Sostituzione di un atomo o di un gruppo di atomi di una molecola con un altro atomo o gruppo di atomi
Reazione endoergonica: Reazione con variazione positiva di energia libera, che pertanto non è spontanea
Reazione endotermica: Reazione in cui si ha assorbimento di calore, pertanto con variazione positiva di entalpia
Reazione esoergonica: Reazione con variazione negativa di energia libera, che pertanto è spontanea
Reazione esotermica: Reazione in cui si ha emissione di calore, pertanto con variazione negativa di entalpia
Reazione intermolecolare: Una reazione che avviene tra due molecole
Reazione intramolecolare: Una reazione che avviene entro la stessa molecola
Reazione monomolecolare: Una reazione in cui la concentrazione di un solo reagente compare nell’espressione della velocità della reazione. Una reazione monomolecolare ha un solo reagente coinvolto nel passaggio che determina la velocità di reazione
Reazione polare: Reazione in cui i legami si formano quando un nucleofilo dona due elettroni ad un elettrofilo, e in cui i legami si rompono quando un frammento esce con entrambi gli elettroni del legame
Reazione radicalica: Reazione in cui i legami vengono formati per donazione di un solo elettrone da ciascuno dei due reagenti, e in cui i legami vengono rotti quando ciascun frammento esce con un solo elettrone
Reazione regioselettiva: Reazione che, dal punto di vista dell’orientamento, dà prevalentemente un solo isomero costituzionale, quando è possibile ottenerne due o più
Reazione regiospecifica: Reazione che, dal punto di vista dell’orientamento, dà esclusivamente un solo isomero costituzionale, quando è possibile ottenerne due o più
Reazione stereoselettiva: Una reazione in cui si produce in maniera preponderante un solo stereoisomero, quando se ne possono formare due o più
Reazione stereospecifica: Una reazione in cui si produce in maniera esclusiva un solo stereoisomero, quando se ne possono formare due o più
Regola di Markovnikov: Nella somma di un composto H-Z ad un alchene non simmetrico, l’idrogeno si lega al carbonio più ricco di idrogeni; ciò è dovuto al fatto che in tal modo si forma come composto intermedio il carbocatione più stabile
Regola di Saytzeff: Nelle reazioni di eliminazione il prodotto preferito è l’alchene che ha il maggior numero di gruppi alchilici legati agli atomi di carbonio impegnati nel doppio legame
Regole di sequenza di Cahn-Ingold-Prelog: Serie di regole per assegnare le priorità relative a gruppi di sostituenti su un atomo di carbonio con doppio legame, su un centro chirale o su un cicloalcano bisostituito
Regole per l’aromaticità di Hückel: Regole che stabiliscono che un composto è aromatico se (1) è ciclico, (2) ha un orbitale p su ogni atomo del ciclo, (3) è planare o quasi planare cosicché vi è una continua, o quasi continua, sovrapposizione tra tutti gli orbitali p dell’anello, e (4) gli elettroni di tale sistemazione ciclica degli orbitali p sono 4n + 2, dove n = 0, 1, 2, 3 ….
Riduzione: Un processo per cui un composto aumenta il numero dei suoi elettroni. Per un composto organico la riduzione comporta la diminuzione dei legami polari e un aumento dei legami C-H
Risoluzione (di un racemo): Separazione di una miscela racemica nei singoli enantiomeri componenti la miscela
Risonanza: Rappresentazione di un composto per mezzo di due o più strutture che differiscono per la sola distribuzione degli elettroni
Ritenzione di configurazione: Una reazione che coinvolge un centro chirale avviene con ritenzione di configurazione se la stereochimica relativa non cambia dai reagenti ai prodotti
Sale di ammonio quaternario: Composto organico che contiene un atomo di azoto carico positivamente che forma quattro legami di tipo s, bilanciato da un opportuno controione negativo X-, per esempio R4N+X-
Sale di diazonio: Composto del tipo R-N=N+ X- dove X- rappresenta un anione, derivato dalla reazione di un’ammina primaria con acido nitroso
Sale di arendiazonio: Composto del tipo Ar-N=N+ X- dove X- rappresenta un anione, derivato dalla reazione di un’anilina con acido nitroso
Sapone: Sale di un acido carbossilico a lunga catena che si ottiene per idrolisi basica di un triacilglicerolo
Saponificazione: Termine antiquato per l’idrolisi indotta da basi di un estere a dare il sale di un acido carbossilico e un alcool
Saturo: Termine usato per indicare molecole organiche che non contengono legami doppi o tripli
Scheletro carbonioso: Struttura di legami s C-C che costituisce lo scheletro di una molecola organica
Scissione eterolitica: Rottura non simmetrica di un legame, tipica delle reazioni polari, quando solo un frammento esce con entrambi gli elettroni di legame
Scissione omolitica: Rottura simmetrica di un legame, tipica delle reazioni radicaliche; su ciascun frammento prodotto resta un elettrone di legame
Sintesi di Williamson: Metodo per preparare eteri per reazione di uno ione alcossido o fenossido con un alogenuro alchilico
Solfonazione: Sostituzione elettrofila aromatica nella quale un anello del benzene reagisce con SO3 o HSO3+ per dare un acido benzensolfonico
Solfone: Composto dalla struttura generale R-SO2-R
Solfossido: Composto dalla struttura generale R-SO-R
Solfuro: Composto con due sostituenti organici alchilici e/o arilici legati allo stesso atomo di carbonio
Solubilità: Misura che indica quanto un composto si scioglie in un solvente
Soluto: Il componente presente in quantità minore in una soluzione
Solvatazione: Associazione di molecole di solvente attorno a una particella di soluto per stabilizzarla
Solvente: Il componente, che scioglie il soluto, presente in quantità maggiore in una soluzione
Solvente polare aprotico: Solvente polare che non è in grado di formare legami idrogeno intermolecolari perchè non contiene gruppi O-H o N-H
Solvente polare protico: Solvente polare che è in grado di formare legami idrogeno intermolecolari perchè contiene gruppi O-H o N-H
Sostanza otticamente attiva: Sostanza che ruota il piano della luce polarizzata
Sostituente: In una molecola organica, gruppo o ramificazione legati alla catena più lunga e continua di atomi di carbonio
Sostituente attivante la sostituzione elettrofila aromatica: Si classifica un gruppo come attivante se l’anello aromatico cui è legato è più reattivo del benzene
Sostituente disattivante la sostituzione elettrofila aromatica: Si classifica un gruppo come disattivante se l’anello aromatico cui è legato è meno reattivo del benzene
Sostituzione nucleofila alifatica: Reazione in cui un nucleofilo ne sostituisce un altro legato ad un atomo di carbonio saturo
Sostituzione nucleofila acilica: Reazione in cui un nucleofilo attacca un composto carbonilico e sostituisce un gruppo uscente legato al carbonio carbonilico
Sostituzione elettrofila aromatica: Nella maggior parte dei casi, sostituzione di un atomo di idrogeno di un anello aromatico operata da un ente con deficienza elettronica (elettrofilo) che si lega al carbonio dell’anello al posto dell’idrogeno
Sostituzione radicalica: Sostituzione di un atomo di idrogeno legato ad un carbonio con un altro atomo o gruppo di atomi condotta da reagenti radicalici. Ne rappresenta un esempio tipico l’alogenazione degli alcani
Spostamento di idruro: Spostamento di un atomo di idrogeno con la propria coppia di elettroni di legame ad un centro cationico vicino
Stadio cineticamente determinante: Lo stadio più lento nella sequenza di una reazione multistadio
Stato di transizione: Massimo di energia raggiunto per una reazione chimica, mentre i reagenti si convertono nei prodotti. Uno stato di transizione è instabile e non può essere isolato
Stereocentro: Atomo di carbonio a cui sono legati quattro gruppi differenti
Stereochimica: Branca della chimica che si occupa della disposizione tridimensionale degli atomi nelle molecole
Stereochimica anti: In una reazione di addizione anti le due estremità di un doppio legame vengono attaccate da lati differenti. In una reazione di eliminazione anti i due gruppi escono da lati opposti della molecola
Stereochimica sin: In una reazione di addizione sin le due estremità di un doppio legame vengono attaccate dallo stesso lato. In una reazione di eliminazione sin i due gruppi escono dallo stesso lato della molecola
Stereoisomeri: Isomeri che differiscono soltanto per il modo in cui gli atomi sono disposti nello spazio, ma che sono uguali per come sono legati fra di loro
Stereospecifico: Termine che indica che un solo stereoisomeri, invece di una miscela, viene prodotto in una data reazione
Struttura condensata: Concisa rappresentazione della struttura di un composto organico in cui sono scritti tutti gli atomi, ma i legami e i doppietti non di legame sono normalmente omessi. Si usano parentesi per denotare gruppi simili legati allo stesso atomo
Struttura di Lewis: Rappresentazione di una molecola che mostra la posizione dei legami covalenti e gli elettroni non di legame. Nelle strutture di Lewis, gli elettroni di valenza sono rappresentati da puntini e un legame covalente a due elettroni è rappresentato da un trattino
Struttura a segmenti: Rappresentazione schematica di una molecola organica in cui gli atomi di carbonio e gli idrogeni non sono rappresentati. Tutti gli eteroatomi e gli idrogeni ad essi legati vengono disegnati, mentre si assume che alla giunzione di ogni due linee o all’estremità di una linea sia presente un atomo di carbonio
Struttura di Kekulé: Metodo di rappresentazione delle molecole in cui le linee tra gli atomi indicano i legami
Strutture di Kekulé del benzene: Due strutture in risonanza del benzene. Ciascuna struttura contiene un anello a sei termini e tre legami p che si alternano a legami s nell’anello
Strutture di risonanza: Due o più strutture per una molecola che differiscono per la posizione dei legami p e per le coppie elettroniche non di legame. Gli atomi e i legami di tipo s invece non vengono coinvolti
Tautomeria: Processo che converte un tautomero in un altro
Tautomeria cheto-enolica: Rapido equilibrio tra la forma carbonilica e quella enolica di una molecola
Tautomeri: Isomeri strutturali che si convertono l’uno nell’altro, di solito per trasferimento di un protone
Tensione angolare: Tensione introdotta in una molecola quando un angolo di legame viene deformato dal proprio valore ideale
Tensione di eclissamento o tensione torsionale: Energia di tensionamento in una molecola provocata da repulsioni elettroniche tra legami eclissati
Tensione sterica: Un aumento dell’energia in una molecola dovuta alla presenza di atomi tra i gruppi che sono costretti a essere troppo vicini
Terminazione: Lo stadio finale di una reazione radicalica a catena in cui due radicali si combinano formando un nuovo legame. Questo passaggio rimuove radicali dalla miscela di reazione senza generarne di nuovi
Tiolo: Composto con il gruppo funzionale -SH
Trans: Prefisso che indica che i due sostituenti di un ciclo o di un doppio legame C=C sono da parti opposte
Variazione di energia libera di Gibbs: Differenza complessiva di energia tra reagenti e prodotti
Variazione di entalpia: Misura della differenza nell’energia di legame totale tra reagenti e prodotti. è detta anche calore di reazione. In una reazione chimica rappresenta la differenza delle energie di legame tra prodotti e reagenti
Variazione di entropia: Misura della differenza in disordine molecolare tra reagenti e prodotti
Vicinale: Termine riferito ad un tipo di sostituzione 1,2
Vinilico: Termine usato per indicare un sostituente di un doppio legame carbonio-carbonio di un alchene
Fonte:
http://unica2.unica.it/galberti/biotin/didattica/mater_didatt/Glossario.doc
http://unica2.unica.it/galberti/biotin/didattica/mater_didatt/materiali.html
Autore del testo: non indicato nel documento di origine
Chimica organica
Visita la nostra pagina principale
Chimica organica
Appunti, riassunti, nozioni di base, sintesi, schemi, brevi tesine utili anche agli studenti delle scuole medie, scuole superiori e università
Termini d' uso e privacy