
Chimica generale 4
Chimica generale 4
Promozione Elettronica
Come abbiamo appena visto, la formazione di legami stabilizza la molecola al punto che, in alcuni casi, un atomo può assumere configurazioni elettroniche meno stabili che tuttavia gli consentono di formare un maggior numero di legami. La promozione elettronica, ad esempio, è un processo di questo tipo, che consente ad un atomo di trasferire un elettrone da un orbitale superficiale saturo ad un orbitale superficiale vuoto. In questo modo un doppietto viene trasformato in due elettroni spaiati che, condivisi con altri atomi, possono essere utilizzati per formare due ulteriori legami chimici.
E’ il caso del Carbonio che, in quasi tutti i suoi composti promuove un elettrone dall’orbitale saturo 2s ad un orbitale 2p vuoto
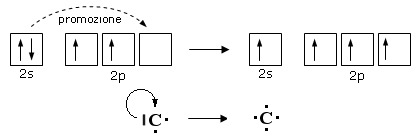
Pur passando da una configurazione elettronica superficiale più stabile ad una meno stabile, il Carbonio dispone ora di 4 elettroni spaiati (contro i due precedenti) che può condividere formando 4 legami chimici.
La promozione elettronica avviene quando la differenza di energia tra l’orbitale di partenza e quello di arrivo è piccola.
La geometria delle molecole: teoria VSEPR
I legami covalenti sono direzionali, nel senso che essi formano tra loro angoli caratteristici che determinano la geometria della molecola. La geometria di una molecola e di conseguenza gli angoli di legame possono essere previsti in modo semplice applicando la teoria VSEPR (Valence-Shell Electron-Pairs Repulsion = repulsione tra doppietti elettronici dello strato di valenza).
Secondo tale teoria i doppietti elettronici più esterni (strato di valenza), essendo carichi negativamente, si respingono, tendendo a disporsi il più lontano possibile gli uni dagli altri, in modo da rendere minima la forza repulsiva e più stabile l'intera molecola.
La teoria prevede inoltre che i doppietti solitari (non impegnati in legami) tendano ad occupare un volume maggiore rispetto ai doppietti elettronici condivisi (impegnati in legami) ed esercitino pertanto una forza repulsiva più intensa. In prima approssimazione possiamo stilare la seguente graduatoria relativa dell'intensità della repulsione esercitata tra coppie di elettroni
repulsione tra coppie solitarie > repulsione tra coppie solitarie e coppie di legame > repulsione tra coppie di legame
Inoltre nella teoria VSEPR i legami doppi e tripli vengono considerati alla stregua di legami semplici e la geometria di una molecola dipende unicamente dal numero di legami (indifferentemente semplici, doppi o tripli) e di coppie solitarie che presenta l’atomo centrale (numero sterico)
numero sterico = numero legami + numero coppie solitarie
La geometria di una molecola è determinata dal suo numero sterico (NS)
- NS=2 - Geometria lineare (AX2)
Molecole con due soli legami e nessun doppietto solitario (AX2) risultano lineari, con le coppie di legame che, respingendosi, si dispongono equidistanti, formando angoli di legame di 180°
X―A―X
Come abbiamo detto, i legami possono essere indifferentemente singoli, doppi o tripli. Presentano, ad esempio, geometria lineare l’idruro di Berillio (BH2), l’anidride carbonica (CO2) e l’acido Cianidrico (HCN)
H―Be―H O=C=O H―C ≡ N
- NS=3 - Geometria trigonale planare (AX3, AX2E)
- Molecole con tre legami e nessun doppietto solitario (AX3) risultano trigonali planari, con le coppie di legame disposte equidistanti su di un piano, con angoli di legame di 120°. Presentano, ad esempio, geometria trigonale planare il cloruro di Boro (BCl3) e l’acetone (H2CO).

- Molecole con due legami ed un doppietto solitario (AX2E) risultano angolate, con un angolo di legame leggermente inferiore a 120° a causa della maggior repulsione del doppietto solitario sui doppietti di legame. Presenta una geometria angolata (derivata da una trigonale planare) l’anidride solforosa (SO2)

- NS=4 – Geometria tetraedrica (AX4, AX3E, AX2E2)
- Molecole con quattro legami e nessun doppietto solitario (AX4) risultano tetraedriche, con le coppie di legame disposte equidistanti ed angoli di legame di 109,5°. E’ il caso del metano (CH4). la cui molecola, come tutte le molecole tridimensionali, può essere rappresentata con legami a cuneo. Si utilizzano cunei pieni per rappresentare i legami che escono dal piano avvicinandosi all’osservatore e cunei tratteggiati per rappresentare i legami che si allontanano.

- Molecole con tre legami ed un doppietto solitario (AX3E) presentano una geometria piramidale di derivazione tetraedrica, con la coppia solitaria ad un vertice del tetraedro che comprime gli angoli di legame, portandoli ad un valore inferiore rispetto a quello caratteristico della geometria tetraedrica. E’ il caso dell’ammoniaca (NH3). la cui molecola piramidale presenta angoli di legame di circa 107°.


- Molecole con due legami e due doppietti solitari (AX2E2) presentano una geometria angolata di derivazione tetraedrica, con le due coppie solitarie ai due vertici del tetraedro che esercitano una forte repulsione e comprimono l’angolo di legame, portandolo ad un valore inferiore rispetto a quello caratteristico della geometria tetraedrica. E’ il caso dell’acqua (H2O). la cui molecola angolata presenta un angolo di legame di 104,5°.

- NS=5 – Geometria bipiramidale trigonale (AX5, AX4E, AX3E2, AX2E3)
- Molecole con cinque legami e nessun doppietto solitario (AX5) risultano bipiramidali trigonali, con tre legami che si dispongono su di un piano (legami equatoriali) a 120° l’uno dall’altro e gli altri due legami (legami assiali) disposti perpendicolarmente, uno sopra e l’altro sotto al piano equatoriale, a formare due piramidi a base triangolare unite per la base. E’ il caso del Pentacloruro di Fosforo (PCl5).

- NS=6 – Geometria ottaedrica (AX6, AX5E, AX4E2, AX3E3, AX2E4)
- Molecole con sei legami e nessun doppietto solitario (AX6) risultano ottaedriche con quattro legami equatoriali distanziati di 90° e due legami equatoriali. Presenta questa geometria l’Esafluoruro di Zolfo (SF6).

- NS=7 – Geometria bipiramidale pentagonale (AX7, AX6E, AX5E2, AX4E3, AX3E4, AX2E5)
- Molecole con sette legami e nessun doppietto solitario (AX7) risultano bipiramidali con cinque legami equatoriali distanziati di 72° e due legami equatoriali. Presenta questa geometria l’Eptafluoruro di Iodio (IF7).

Naturalmente in tutte le strutture, l’eventuale presenza di doppietti solitari modifica la geometria originaria, comprimendo gli angoli dei legami residui.
Geometrie VSEPR |
|||||
|
Coppie solitarie |
||||
0 |
1 |
2 |
3 |
4 |
|
NS=2 |
|
|
|
|
|
NS=3 |
|
|
|
|
|
NS=4 |
|
|
|
|
|
NS=5 |
|
|
|
|
|
NS=6 |
|
|
|
|
|















Legame covalente polare: elettronegatività e momento di dipolo
Quando gli elettroni vengono condivisi da atomi del medesimo elemento, ciascun atomo li attrae con la medesima intensità. In questo caso gli elettroni condivisi (elettroni di legame) possono essere immaginati come una nuvola negativa che si dispone in maniera uniforme e simmetrica intorno ai due nuclei senza produrre alcun tipo di polarità sulla molecola. Si parla in questo caso di legame covalente puro.
Nella maggior parte dei casi però gli atomi che formano il legame covalente appartengono ad elementi diversi che presentano una diversa forza di attrazione sugli elettroni di legame.
Si definisce elettronegatività χ (la lettera greca “chi”) la forza con cui un atomo attira a sé gli elettroni condivisi.
L'elettronegatività è una grandezza di difficile valutazione poiché, a differenza dell'affinità elettronica e dell'energia di ionizzazione che si riferiscono ad atomi isolati, l’elettronegatività si riferisce ad atomi legati ad altri atomi.
In generale il valore dell'elettronegatività può dunque variare, per uno stesso elemento, in relazione al tipo e al numero di atomi di altri elementi impegnati nel legame.
Nonostante ciò, al fine di avere a disposizione un parametro che permetta di valutare, anche se in modo approssimato, la polarità di un legame, sono stati proposti diversi metodi di calcolo per assegnare un valore di elettronegatività ai diversi elementi.
Tra i metodi più importanti vi sono quelli proposti da Mulliken e da Pauling.
L'elettronegatività secondo Mulliken è pari alla media aritmetica dell'energia di ionizzazione e dell'affinità elettronica.

L'elettronegatività secondo Pauling di un elemento A viene calcolata conoscendo l'elettronegatività di un elemento B attraverso la seguente relazione

Pauling ammette cioè che la differenza di elettronegatività tra due elementi sia uguale alla radice quadrata di una quantità Δ, detta energia di risonanza ionico-covalente espressa in eV, il cui valore è dato da

dove
DAB = energia di legame del composto A-B
DAA = energia di legame del composto A-A
DBB = energia di legame del composto B-B
 = media geometrica delle energie dei legami covalenti puri A-A e B-B, assunta come stima dell’energia di legame di un ipotetico legame covalente puro A-B
= media geometrica delle energie dei legami covalenti puri A-A e B-B, assunta come stima dell’energia di legame di un ipotetico legame covalente puro A-B
.
In altre parole, l'energia di risonanza ionico-covalente Δ misura la differenza di energia tra il legame covalente reale AB ed un ipotetico legame covalente puro AB.
Nel caso l'energia di legame sia espressa in kJ/mol o in kcal/mol è necessario applicare un coefficiente k di conversione (per trasformare in eV/particella), che vale rispettivamente 0,0103643 e 0,0433641.

Per poter utilizzare la relazione di Pauling è evidentemente necessario fissare arbitrariamente l'elettron egatività di un elemento che faccia da riferimento. Pauling assunse per l'idrogeno χ = 2,1.
Esempio L'energia del legame HCl considerato come covalente puro è pari a La differenza di elettronegatività calcolata è pertanto
Sapendo che l'elettronegatività dell'idrogeno è convenzionalmente 2,1 si ottiene per il Cloro |




La scala di Mulliken è più rigorosa della scala di Pauling essendo costruita su grandezze misurabili. Nella pratica si usa però prevalentemente la scala di Pauling in quanto per molti elementi il valore dell'affinità elettronica è di difficile determinazione.
D'altra parte le due scale forniscono valori in gran parte coincidenti, risultando legate, anche se in modo approssimato, dalla seguente relazione

Esempio |


I valori di elettronegatività secondo Pauling si trovano tabulati nella tabella periodica e presentano il valore minimo in basso a sinistra (Francio = 0.7) e crescono diagonalmente fino ad assumere il valore massimo in alto a destra (Fluoro = 4).
Elettronegatività (Pauling) |
|||||||||||||||||
H |
|
He |
|||||||||||||||
Li |
Be |
|
B |
C |
N |
O |
F |
Ne |
|||||||||
Na |
Mg |
|
Al |
Si |
P |
S |
Cl |
Ar |
|||||||||
K |
Ca |
Sc |
Ti |
V |
Cr |
Mn |
Fe |
Co |
Ni |
Cu |
Zn |
Ga |
Ge |
As |
Se |
Br |
Kr |
Rb |
Sr |
Y |
Zr |
Nb |
Mo |
Tc |
Ru |
Rh |
Pd |
Ag |
Cd |
In |
Sn |
Sb |
Te |
I |
Xe |
Cs |
Ba |
La |
Hf |
Ta |
W |
Re |
Os |
Ir |
Pt |
Au |
Hg |
Tl |
Pb |
Bi |
Po |
At |
Rn |
Fr |
Ra |
Ac |
Rf |
Db |
Sg |
Bh |
Hs |
Mt |
Ds |
Rg |
Uub |
Uut |
Uuq |
Uup |
Uuh |
Uus |
Uuo |
Ce |
Pr |
Nd |
Pm |
Sm |
Eu |
Gd |
Tb |
Dy |
Ho |
Er |
Tm |
Yb |
Lu |
Th |
Pa |
U |
Np |
Pu |
Am |
Cm |
Bk |
Cf |
Es |
Fm |
Md |
No |
Lr |
E' evidente che tanto maggiore sarà la differenza di elettronegatività tra due elementi impegnati in un legame, tanto maggiore sarà la polarità del legame.
Dunque, quando si forma un legame covalente tra due atomi che presentano diversa elettronegatività la nube elettronica che costituisce il legame covalente risulta spostata verso l'atomo più elettronegativo. Quest'ultimo acquista pertanto una carica parzialmente negativa (indicata con -), mentre l'altro una carica parzialmente positiva (d+). La distribuzione asimmetrica della nuvola elettronica produce dunque due poli aventi carica opposta (dipòlo) ed il legame viene perciò definito covalente polare.
E’ ciò che accade, ad esempio nella molecola dell’acido Cloridrico (HCl) in cui l’Idrogeno condivide un elettrone con il Cloro. Il Cloro più elettronegativo presenta una parziale carica negativa e la molecola di HCl risulta polare

La distribuzione di carica elettrica di un dipolo può essere rappresentata tramite una mappa (o superficie) di potenziale elettrostatico (o densità elettronica) dove le tonalità del rosso indicano la carica negativa, quelle del blu la carica positiva, mentre il verde la neutralità.

Maggiore è la differenza di elettronegatività (Δχ) tra i due elementi e maggiore sarà la polarità del legame (le cariche parziali saranno più vicine ad una intera carica).
Quando la differenza di elettronegatività tra i due elementi supera il valore critico di 1.9, si assume che l’elemento più elettronegativo sia in grado di strappare l’elettrone all’altro elemento ed il legame viene descritto come ionico. Possiamo dunque descrivere il legame ionico come un caso limite del legame covalente polare per Δχ > 1.9
L'intensità di un dipolo si esprime attraverso la determinazione del suo momento dipolare. Si definisce momento dipolare μ il prodotto della carica q associata ad uno dei baricentri di carica (la carica dell'altro baricentro ha valore uguale e di segno opposto) per la distanza r tra i baricentri.
μ = Q • r
L'unità di misura del momento dipolare è il debye (D).
Un momento dipolare presenta l'intensità di 1 debye quando 2 cariche elettriche di segno opposto, aventi intensità di 10-10 u.e.s. (unità elettrostatiche o franklin) si trovano alla distanza di 1 Å.
1 D = 10-10 ues Å = 3,335641·10-30 C m
Per caratterizzare la polarità di un legame covalente è possibile assegnargli una certa percentuale di carattere ionico, calcolabile in funzione del suo momento dipolare.
La percentuale di carattere ionico si calcola come rapporto percentuale tra il momento dipolare effettivo (misurato) ed il momento dipolare di un teorico legame ionico

Il momento dipolare di un ipotetico legame completamente ionico si calcola con
μionico = Qe• r = 4,8 • r
dove Qe è la carica dell'elettrone (e = 4,8 10-10 u.e.s.) ed r il valore della lunghezza del legame in Å.
Ad esempio, sapendo che il momento dipolare dell’acido cloridrico è μHCl = 1,1 D e la lunghezza del legame H-Cl è di 1,27 Å, si calcola

Sopra l’atomo di Cloro è presente una parziale carica negativa (d-) pari al 18% dell’intera carica dell’elettrone, mentre sopra l’atomo di Idrogeno sarà presente una parziale carica positiva (d+) della medesima intensità.
La polarità di un legame può anche essere stimata utilizzando la relazione di Pauling che correla la percentuale di carattere ionico alla differenza di elettronegatività (Δχ).

Ad esempio, sapendo che la differenza di elettronegatività tra Idrogeno e Fluoro è pari a Δχ = χF – χH = 4 - 2,2 = 1,8, possiamo stimare la percentuale di carattere ionico dell’acido fluoridrico

Il momento dipolare è una grandezza vettoriale, che viene rappresentata con una freccia orientata dal polo positivo al quello negativo.

Nelle molecole in cui sono presenti più legami il momento di dipolo dell’intera molecola risulta essere la somma vettoriale dei momenti di dipolo dei singoli legami. Se il momento di dipolo risultante è diverso da zero allora la molecola è polare. Se il momento di dipolo risultante è uguale a zero, la molecola risulta apolare (anche se i suoi legami sono polari).
La polarità di una molecola dipende quindi non solo dalla polarità dei suoi legami, ma anche dalla sua geometria.
Così, ad esempio, se confrontiamo la polarità dell’anidride carbonica e dell’acqua, troveremo che mentre l’anidride carbonica è apolare, l’acqua è polare. L’anidride carbonica è infatti una molecola lineare ed il momento di dipolo dei suoi due legami risulta essere uguale e contrario, per cui il momento risultante è nullo.
L’acqua è invece una molecola angolata e la polarità dei suoi due legami si compone vettorialmente per dare un momento di dipolo diverso da zero. L’acqua è un dipolo.
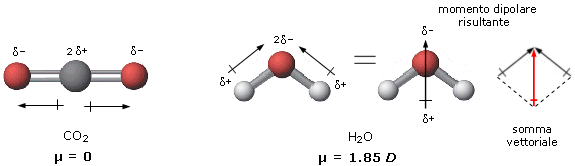
In effetti il momento dipolare totale di una molecola è dato dalla somma vettoriale non solo dei momenti dipolari relativi ai legami covalenti, ma anche ai dipoli associati alle coppie solitarie presenti nella molecola.

Una molecola polare è un minuscolo dipòlo il quale è in grado di ruotare orientandosi opportunamente se posto in un campo elettrico.

Dipoli orientati in un campo elettrico
Molecole con elettroni spaiati e paramagnetismo
Non sempre gli atomi utilizzano tutti i loro elettroni spaiati per effettuare legami chimici. In qualche caso può accadere che in una molecola sopravvivano degli orbitali insaturi.
Un tipico esempio di tale comportamento è rappresentato dal monossido di azoto.

Tutte le sostanze che si trovano a possedere un elettrone spaiato risultano essere paramagnetiche, vengono cioè debolmente attratte dai poli di un magnete. Tale comportamento è dovuto proprio al debole campo magnetico associato all'elettrone, non compensato in questo caso da un elettrone con spin opposto.
Strutture di Lewis molecolari e carica formale
La convenzione introdotta da Lewis per rappresentare gli elettroni di valenza degli elementi viene utilizzata anche per rappresentare intere molecole. Per scrivere la struttura di Lewis di una molecola è necessario conoscere la sua formula molecolare e la connettività.
La formula molecolare e la connettività sono determinate sperimentalmente e devono essere note. La connettività o costituzione è l’ordine con cui gli atomi di una molecola sono connessi.
Ad esempio il nitrito di metile ha formula molecolare CH3NO2 e la sua connettività è C-O-N-O con tutti gli idrogeni legati al carbonio.
Vediamo di seguito i 6 passaggi necessari per scrivere correttamente una struttura di Lewis molecolare. Useremo il nitrito di metile CH3NO2.
- Determinare il numero degli elettroni di valenza della molecola.
Per una molecola neutra, il numero di elettroni di valenza è uguale alla somma degli elettroni di valenza degli atomi coinvolti (elettroni superficiali che compaiono nella struttura di Lewis dell’elemento). Si ricordi che il numero di elettroni superficiali coincide in genere con il numero d’ordine del gruppo chimico al quale l’elemento appartiene. Così l’Ossigeno (VI gruppo A) possiede 6 elettroni di valenza, l’Azoto (V gruppo A) ne possiede 5, il Carbonio 4 (IV gruppo A) e l’Idrogeno 1 elettrone di valenza (I gruppo A).
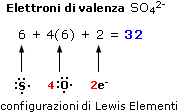
Per una molecola elettricamente carica il numero di elettroni di valenza è uguale al numero di elettroni di valenza degli atomi, al quale va sommato il numero delle cariche negative o sottratto il numero delle cariche positive. Ad esempio l’anione solfato SO42- possiede 32 elettroni di valenza (6 per l’atomo di Zolfo, 6 per ogni atomo di ossigeno e 2 per le due cariche negative dell’anione).
Il nitrito di metile (CH3NO2) presenta 24 elettroni di valenza. Ogni idrogeno contribuisce infatti con 1 elettrone di valenza, il carbonio con 4, l’azoto con 5 ed ogni atomo di ossigeno con 6 per un totale di 24 elettroni.

- Costruire un primo schema di legame, rispettando la connettività e collegando gli atomi con un legame covalente semplice.

- Determinare gli elettroni residui da posizionare. Sottrarre gli elettroni di legame dagli elettroni di valenza, ottenendo in tal modo il numero di elettroni che devono ancora essere posizionati. Nel nitrito di metile CH3NO2, gli elettroni di legame sono 12, mentre quelli di valenza sono 24 e devono pertanto essere ancora posizionati 24 – 12 = 12 elettroni.
- Aggiungere gli elettroni residui come coppie di elettroni di non-legame (elettroni non condivisi o coppie solitarie o lone pairs) in modo che il maggior numero di atomi presenti 8 elettroni (ovviamente non l’idrogeno), iniziando con gli atomi più elettronegativi.

- Spostare coppie solitarie per completare l’ottetto. Se un atomo non ha l’ottetto completo, usare una coppia di elettroni solitari dell’atomo adiacente che possiede il maggior numero di doppietti non condivisi, per formare un doppio o un triplo legame. Nella struttura precedente si osserva, ad esempio, che l’azoto ha solo 6 elettroni (4 condivisi con i due atomi di Ossigeno e 2 non condivisi). Trasferiamo dunque un doppietto solitario dall’Ossigeno terminale (che ha 3 doppietti non condivisi, contro i 2 doppietti dell’ossigeno centrale), per formare un doppio legame N=O.

La regola per la quale ogni atomo legato deve avere non più di 8 elettroni superficiali (regola dell’ottetto) vale rigorosamente solo per gli elementi non metallici del secondo periodo chimico (C N O F) i quali, non possedendo orbitali d, non sono in grado di trasferirvi elettroni (promozione elettronica) per aumentare il numero dei loro legami. I primi elementi del secondo periodo possono avere meno di otto elettroni nel loro stato legato (4 per il Berillio e 6 per il Boro = ottetto incompleto). Gli elementi non metallici dei periodi superiori al secondo possono invece avere più di 8 elettroni superficiali (ottetto espanso) nel loro stato legato. In particolare quelli del gruppo V-A, come il Fosforo, possono avere 10 elettroni superficiali, mentre quelli del gruppo VI-A come lo Zolfo possono arrivare a 12 e quelli del gruppo VII-A, come il Cloro ne possono avere 14.
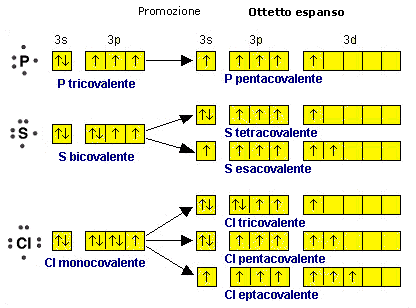
Ad esempio Lo Zolfo completa l’ottetto nell’acido solfidrico, mentre arriva a 10 elettroni nell’acido solforoso e a 12 elettroni nell’acido solforico

- Calcolare la carica formale di ciascun atomo. La carica atomica formale viene definita come la carica che un atomo avrebbe se tutti i suoi legami venissero considerati come covalenti puri. La carica formale di ciascun atomo e' data dunque dalla differenza tra gli elettroni di valenza dell'atomo isolato e gli elettroni di valenza dell'atomo legato nella molecola. Per determinare gli elettroni di valenza dell’atomo legato (elettroni che l’atomo possiede nella molecola) è necessario assegnargli un elettrone per ogni coppia di elettroni di legame ed entrambi gli elettroni di ogni sua coppia solitaria. Si confrontano poi gli elettroni di valenza dell’atomo legato con gli elettroni di valenza dell’atomo isolato neutro. Ogni elettrone in eccesso rappresenta una carica formale negativa. Ogni elettrone in difetto una carica formale positiva.
carica formale = elettroni di valenza atomo isolato – elettroni di valenza atomo legato
carica formale = elettroni di valenza – [½ elettroni di legame + elettroni solitari]
Naturalmente la somma delle cariche formali di tutti gli atomi di una molecola o ione deve essere uguale alla carica elettrica complessiva della molecola.
Per calcolare la carica formale sostituiamo i trattini che rappresentano i legami con coppie di puntini (i due elettroni condivisi) e successivamente contiamo gli elettroni intorno ai singoli atomi confrontandoli con i rispettivi elettroni di valenza degli atomi isolati

Intorno a ciascun atomo di ossigeno vi sono 6 elettroni. Poiché l’atomo di ossigeno isolato possiede 6 elettroni superficiali, i due atomi di ossigeno presentano carica formale nulla (6-6=0)

Intorno all’atomo di carbonio vi sono 4 elettroni, mentre intorno all’atomo di azoto vi sono 5 elettroni. Poiché gli atomi di carbonio e di azoto isolati possiedono rispettivamente 4 e 5 elettroni superficiali, la loro carica formale è nulla.

Esaminiamo ora il nitrometano, un composto che presenta la medesima formula molecolare del nitrito di metile CH3NO2, ma diversa connettività o costituzione (nitrometano e nitrito di metile sono due isomeri costituzionali). Nel nitrometano il carbonio si lega direttamente al’azoto il quale si lega ai due atomi di ossigeno. Il primo schema di legame è dunque il seguente

Abbiamo posizionato 6 legami per un totale di 12 elettroni. Gli elettroni di valenza sono sempre 24, come per il nitrito di metile (gli atomi sono gli stessi) e ci rimangono dunque altri 12 elettroni da posizionare. Assegnamo 3 doppietti solitari a ciascun atomo di ossigeno (più elettronegativo dell’azoto) in modo da raggiungere una configurazione ad 8 elettroni

In questo modo l’azoto non presenta tuttavia l’ottetto completo (ha solo 3 legami per un totale di 6 elettroni) e quindi trasferiamo un doppietto solitario di uno dei due atomi di Ossigeno per formare un doppio legame N=O

Calcoliamo ora la carica formale di ciascun atomo. Sostituiamo i trattini che rappresentano i legami con coppie di puntini (i due elettroni condivisi) e successivamente contiamo gli elettroni intorno ai singoli atomi (elettroni di valenza degli atomi legati)

L’atomo di ossigeno con il doppio legame presenta 6 elettroni, di cui 2 elettroni per i due legami e 4 per i due doppietti solitari). Avendo l’ossigeno isolato 6 elettroni di valenza la sua carica formale è nulla. L’atomo di ossigeno con il legame semplice presenta invece 7 elettroni, di cui 1 elettrone per il legame e 6 per i tre doppietti solitari. Avendo dunque un elettrone in più rispetto ai 6 elettroni di valenza di un atomo di ossigeno isolato, la sua carica formale è -1.

L’atomo di Azoto presenta 4 elettroni (metà degli 8 elettroni che formano i suoi 4 legami) Avendo dunque un elettrone in meno rispetto ai 5 elettroni di valenza di un atomo di Azoto isolato, la sua carica formale è +1.

L’atomo di Carbonio presenta 4 elettroni (metà degli 8 elettroni che formano i suoi 4 legami) Avendo dunque il medesimo numero di elettroni di un atomo di carbonio isolato, la sua carica formale è nulla.

La formula di Lewis del nitrometano sarà pertanto

Si rammenti che una formula di Lewis non è completa se non presenta le corrette cariche formali.
Il Legame covalente: Teoria del legame di valenza (VB)
La teoria del legame di valenza (Valence Bond Theory) fu proposta nel 1927 da W.Heitler e F.London e successivamente ampliata e sviluppata da L.Pauling con l’introduzione dei concetti di risonanza (1928) e di ibridazione orbitalica (1930). La teoria interpreta la formazione del legame covalente mediante il concetto quantomeccanico di orbitale.
Il legame covalente, che nella teoria di Lewis viene visto come una condivisione da parte di due atomi di una coppia di elettroni, viene descritto come una sovrapposizione degli orbitali atomici che ospitano i due elettroni spaiati da condividere.
Le funzioni d’onda dei due orbitali si sommano (in modo analogo ai fenomeni di interferenza per le onde meccaniche) per dare una nuova funzione d’onda che descrive un nuovo orbitale.
Il nuovo orbitale appartiene ad entrambi gli atomi legati ed ospita i due elettroni con spin antiparallelo.
Nel caso del legame covalente semplice che tiene uniti due atomi di Idrogeno nella molecola H2, ad esempio, abbiamo una sovrapposizione di due orbitali 1s. Se indichiamo i due atomi di Idrogeno con HA e HB, le due funzioni d’onda che si sommano sono ΨA(1s) e ΨB(1s)
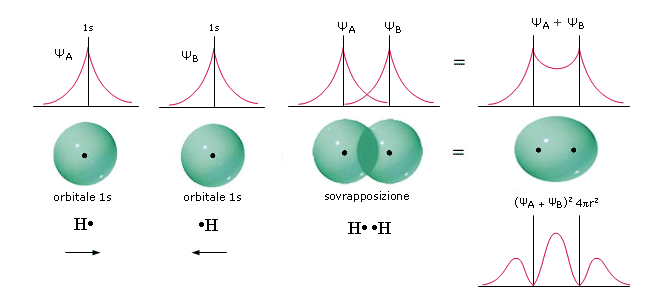
La funzione di distribuzione radiale della densità elettronica (probabilità) del nuovo orbitale che si è formato mostra un massimo tra i due nuclei. Si suppone che, quando gli atomi di H si avvicinano, ciascun elettrone condiviso possa passare da un nucleo all’altro, cioè che a distanze ravvicinate i nuclei non ‘distinguano’ gli elettroni di legame.
Nel formare i legami gli orbitali, se possibile, tendono a massimizzare la regione di sovrapposizione. Gli orbitali di tipo p, ad esempio, tendono a sovrapporsi lungo il loro asse maggiore. Nella molecola biatomica del Fluoro F2, ad esempio, due orbitali 2p si sovrappongono lungo l’asse maggiore, utilizzando i lobi aventi il medesimo segno, in modo che la funzione d’onda tra i due nuclei si rinforzi, aumentando la densità elettronica.

Questo tipo di sovrapposizione genera un legame covalente particolarmente intenso, detto legame σ.
Nel caso di legami covalenti doppi e tripli, solo una coppia di orbitali p può generare un legame σ. Gli altri orbitali p, essendo disposti uno perpendicolarmente all’altro, sono costretti a sovrapporsi lateralmente (lungo l’asse minore). Questo tipo di legame covalente è più debole (a causa della minor sovrapposizione) ed è detto legame π.

Quando in una molecola si forma un legame covalente doppio si genera un legame σ lungo la congiungente i due nuclei ed un legame π costituito da due nuvole elettroniche disposte simmetricamente (sopra e sotto) rispetto al legame σ. Un doppio legame è una struttura rigida e non consente la libera rotazione dei due atomi legati attorno all’asse di legame.

Il legame doppio è quindi più forte di un legame semplice, ma presenta tuttavia una forza inferiore a quella di due legami semplici essendo costituito da un legame σ (più forte) ed un legame π (più debole).
Quando in una molecola si forma un legame covalente triplo si genera un legame σ lungo la congiungente i due nuclei e due legami π costituiti da quattro nuvole elettroniche disposte simmetricamente ai quattro lati del legame σ (un legame sopra-sotto ed un legame davanti-dietro). Anche un triplo legame è una struttura rigida e non consente la libera rotazione dei due atomi legati attorno all’asse di legame.

In una molecola biatomica come l’azoto (N2), ad esempio, in cui due atomi di Azoto sovrappongono tre coppie di orbitali p formando un legame covalente triplo, gli orbitali px si compenetrano lungo la congiungente i due nuclei formando un legame di tipo σ, mentre gli altri orbitali p si sovrappongono lateralmente dando origine a due legamiπ che presentano un massimo di densità elettronica sopra e sotto l’asse internucleare.
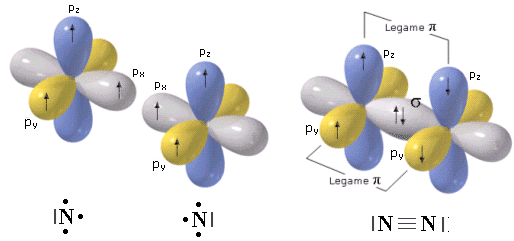
Il legame triplo è quindi più forte di un legame semplice, ma presenta tuttavia una forza inferiore a quella di tre legami semplici essendo costituito da un legame σ (più forte) e da due legami π (più deboli).
In definitiva, nel caso in cui la densità elettronica si concentri sull’asse internucleare, si parla di legameσ, nel caso si concentri sopra e sotto l’asse internucleare si parla di legameπ.
I legami σ presentano una simmetria cilindrica e sono quindi invarianti per rotazione attorno all’asse di legame.
I legami π non sono cilindricamente simmetrici rispetto all'asse di legame, poiché la funzione d’onda cambia di segno per rotazione attorno all’asse.

Presentano simmetria σ anche i legami che si formano per sovrapposizione di due orbitali s, come nella molecola H2, o per sovrapposizione di un orbitale s con un orbitale p, come nella molecola HCl.

Ibridazione orbitalica
Per formare legami, gli atomi possono ricombinare gli orbitali atomici (s,p,d) per dar luogo ad un ugual numero di orbitali atomici detti orbitali ibridi. Questo processo, detto ibridazione, è un procedimento di combinazione matematica delle funzioni d’onda originarie.
L’ibridazione interessa orbitali superficiali (di valenza) con contenuto energetico non molto diverso. Gli orbitali ibridi così formati sono tutti di uguale forma ed energia e sono orientati in modo da interferire il meno possibile fra di loro, massimizzando la reciproca distanza.
Gli orbitali ibridi più importanti sono quelli che si formano dalla combinazione di un orbitale s con uno o più orbitali p. La superficie di contorno di tali orbitali ibridi è costituita da due lobi contrapposti di diversa dimensione in cui la funzione d’onda Ψ assume segno opposto. Il lobo di dimensione maggiore è quello che viene utilizzato nei legami.

La combinazione di un orbitale di tipo s e uno di tipo p dà origine a due orbitali ibridi detti orbitali sp
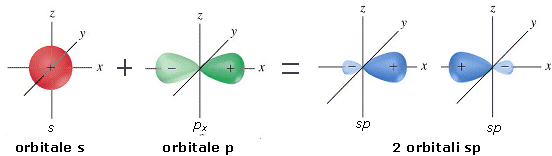
Gli orbitali sp si dispongono a 180° l’uno rispetto all’altro. Nella visione d’insieme spesso si omette di rappresentare il lobo minore di ciascun orbitale ibrido.

Gli orbitali p non ibridati si orientano perpendicolarmente alla retta di ibridazione e perpendicolarmente tra loro.

L’ibridazione sp è tipica di molecole con una geometria lineare. Presentano un’ibridazione sp l’atomo di Berillio nell’idruro di Berillio (BH2), l’atomo di carbonio nell’anidride carbonica (CO2) e gli atomi di carbonio uniti da un legame covalente triplo (-C≡C-) come ad esempio nella molecola dell’etino HC≡CH

Etino
Va detto che, per semplicità, si disegnano normalmente gli ibridi con il piano nodale passante per il nucleo, mentre il nucleo si trova in una zona a densità elettronica non nulla.

La combinazione di un orbitale di tipo s e di due orbitali di tipo p dà origine a tre orbitali ibridi detti orbitali sp2 che si dispongono su di un piano a 120° l’uno dall’altro.

L’orbitale p non ibridato si dispone perpendicolarmente al piano di ibridazione.

L’ibridazione sp2 è tipica di molecole con una geometria trigonale planare. Presentano un’ibridazione sp2 l’atomo di Boro nel cloruro di Boro (BCl3) e gli atomi di carbonio uniti da un legame covalente doppio (>C=C<), come ad esempio nella molecola dell’etene (o etilene) H2C=CH2

Etene
La combinazione di un orbitale di tipo s e di tre orbitali di tipo p dà origine a quattro orbitali ibridi detti orbitali sp3 che puntano verso i vertici di un tetraedro, disponendosi a 109,5° l’uno dall’altro.

L’ibridazione sp3 è tipica di molecole con una geometria tetraedrica. Presenta un’ibridazione sp3 l’atomo di Carbonio nel metano (CH4) ed in tutti i casi in cui forma quattro legami covalenti semplici.

Metano
Sono possibili ibridazioni più complesse che coinvolgono anche gli orbidali d e che corrispondono alle geometrie già studiate con la teoria VSEPR.
Ibridazione |
Geometria |
Molecola |
sp |
lineare |
|
sp2 |
Trigonale planare |
|
sp3 |
Tetraedrica |
|
sp3d |
Bipiramidale trigonale |
|
sp3d2 |
Ottaedrica |
|
sp3d3 |
Bipiramidale pentagonale |
|






Risonanza o mesomeria
La risonanza è un concetto quantomeccanico introdotto da Pauling per descrivere lo stato di legame di una molecola, altrimenti non descrivibile attraverso una normale formula di struttura. Per descrivere la molecola si utilizzano pertanto più formule di struttura, dette formule-limite (o strutture contribuenti o formule-canoniche o strutture di risonanza), aventi la medesima connettività, ma diversa disposizione degli elettroni superficiali. Le diverse strutture-limite si differenziano solo per la posizione dei legami multipli (elettroni π) e dei doppietti solitari.. La molecola reale è detta ibrido di risonanza ed avrà una configurazione elettronica intermedia tra quelle delle formule-limite.
Consideriamo, ad esempio, la molecola dell'anidride solforosa SO2, alla quale avevamo già assegnato una formula di struttura del tipo

dove lo zolfo si lega ad un atomo di ossigeno con legame singolo (dativo) e ad un altro con legame doppio. Ci dobbiamo dunque attendere una molecola asimmetrica, con un legame leggermente più lungo (legame semplice) ed uno più corto (legame doppio).
In realtà le osservazioni sperimentali indicano che la molecola dell’anidride solforosa è perfettamente simmetrica ed i due legami hanno esattamente la stessa lunghezza, la quale risulta essere intermedia tra quella di un legame semplice e quella di un legame doppio. Poiché non è tuttavia possibile rappresentare la reale struttura dell’anidride solforosa con una unica formula si utilizzano più formule di struttura. Nel caso specifico dell’anidride solforosa ne sono sufficienti due.
Le strutture-limite vanno separate da una freccia a due punte e nessuna di esse da sola è in grado di descrivere correttamente la molecola reale. Il movimento degli elettroni per passare da una struttura all’altra viene rappresentato utilizzando frecce curve. Gli unici movimenti elettronici consentiti sono i seguenti
- Da doppietto solitario a legame adiacente per formare un legame multiplo (doppio o triplo) o viceversa

- Da legame multiplo a legame adiacente per spostare elettroni π

Come in tutte le strutture di Lewis, anche nel caso delle strutture risonanti le cariche formali vanno indicate.
Per l’anidride solforosa avremo dunque

La struttura reale dell’anidride solforosa è intermedia tra le due strutture limite o, come si suol dire, è un ibrido di risonanza.
La risonanza si produce poiché l'ibrido risulta energeticamente favorito ed è quindi più stabile di ognuna delle strutture limite che ad esso contribuiscono.
Le strutture-limite possono contribuire in misura diversa all’ibrido in relazione al loro contenuto energetico.
Le strutture-limite più stabili contribuiscono maggiormente all’ibrido. Questo significa che l’ibrido assomiglierà maggiormente, sia come struttura che come contenuto energetico alla struttura-limite più stabile. In altre parole l’ibrido è una media ponderata delle sue strutture-limite ed il fattore di ponderazione è la stabilità di ciascuna struttura.
Viene definita energia di risonanza la differenza tra l’energia della molecola reale (misurata) e quella della sua struttura di risonanza più stabile (calcolata).
Un ibrido di risonanza è tanto più stabile (elevata energia di risonanza) quanto più numerose e tra loro equivalenti dal punto di vista energetico sono le sue strutture-limite.
E' allora evidente che se una delle strutture-limite risulta molto più stabile di tutte le altre, l'ibrido assomiglierà a tal punto a quest'ultima da rendere la risonanza poco evidente e si potrà pertanto accettare la struttura più stabile come una buona approssimazione della reale struttura molecolare.

Nel caso dell'anidride solforosa, ad esempio, le due strutture limite sono perfettamente simmetriche ed energeticamente equivalenti, per cui il fenomeno della risonanza sarà particolarmente accentuato.
Nel caso dell'anidride carbonica troviamo invece un esempio di una struttura limite leggermente più stabile delle altre.
L'anidride carbonica viene normalmente rappresentata con la seguente formula di struttura

Si rileva però sperimentalmente che i due legami Carbonio-Ossigeno presentano una lunghezza intermedia tra quella di un legame doppio e quella di un legame triplo. Inoltre, sapendo che quando si forma un doppio legame C=O si liberano 175 Kcal/mol, ci si attende che la formazione di una mole di CO2 a partire da C e O2, liberi circa 350 Kcal. Il calore di formazione misurato sperimentalmente per l'anidride carbonica è invece di 383 Kcal/mol.
Evidentemente l'anidride carbonica risulta essere più stabile di quanto non ci si attendesse sulla base di una struttura ipotizzata del tipo O=C=O. Tale aumento di stabilità deve essere attribuito al fatto che l'anidride carbonica è in realtà un ibrido di risonanza. La differenza energetica di 33 Kcal/mol rappresenta l'energia di risonanza.
Si attribuiscono all'anidride carbonica le seguenti strutture limite

Le strutture non sono egualmente stabili. La struttura centrale, senza cariche formali, risulta essere più stabile e contribuisce in maggior misura all'ibrido. La struttura reale assomiglia di più ad essa di quanto non assomigli alle altre due strutture.
Delocalizzazione elettronica
Da un punto di vista fisico la risonanza è essenzialmente un modo per descrivere un fenomeno di delocalizzazione che interessa i sistemi coniugati.
Un sistema coniugato è costituito da un orbitale p su di un atomo adiacente ad un legame π (legame doppio o triplo). Vi sono 4 possibili configurazioni per un sistema coniugato:
1) l’orbitale p è coinvolto anch’esso in un legame π (doppi legami coniugati)
– X = Y – Z = W–
2) L’orbitale p è vuoto (tipicamente, ma non necessariamente, l’atomo che lo porta è carico positivamente)
– X = Y – Z +
3) L’orbitale p è saturo (tipicamente, ma non necessariamente, l’atomo che lo porta è carico negativamente)
– X = Y – Z:–
4) L’orbitale p è semisaturo (radicale coniugato)
– X = Y – Z ·
Quando si presenta una di queste configurazioni, vi sono le condizioni affinché si produca un fenomeno di delocalizzazione elettronica (rappresentabile attraverso strutture di risonanza) tra gli atomi del sistema coniugato. I due orbitali p del doppio legame e l’orbitale p dell’atomo adiacente risultano infatti sovrapposti portando ad una delocalizzazione degli elettroni su 3 atomi (4, nel caso di doppi legami coniugati). Tutti gli atomi coinvolti nella delocalizzazione risultano ibridati sp2 (o sp) e quindi planari, con gli orbitali p disposti perpendicolarmente al piano d’ibridazione e parallelamente l’uno rispetto all’altro. La complanarità degli atomi coinvolti, con gli orbitali p disposti parallelamente, è essenziale affinchè gli orbitali p possano sovrapporsi e dar luogo alla delocalizzazione e quindi alla risonanza.
Se ad esempio analizziamo la struttura di Lewis dell’etenammina senza considerare la risonanza saremmo indotti a ritenere che l’atomo di Azoto sia ibridato sp3 (presenta 3 legami ed un doppietto solitario)

In realtà l’etenammina è una molecola perfettamente planare, come ci suggerisce la sua seconda struttura di risonanza in cui è presente un legame doppio C=N

Anche l’Azoto è dunque ibridato sp2 in modo che il suo orbitale p, contenente il doppietto solitario, sia parallelo agli orbitali p dei due atomi di carbonio e si possa sovrapporre ad essi.

La presenza di gruppi chimici ingombranti che impediscano il parallelismo tra gli orbitali p, inibisce il fenomeno della risonanza (inibizione sterica della risonanza).
In alcuni casi tale condizione può essere graficamente rappresentata senza ricorrere alle formule limite. Ad esempio l'anidride solforosa può essere rappresentata anche così

con gli elettroni π delocalizzati su tutta la molecola indicati dalla linea tratteggiata.
A) Sistema coniugato π- π
Nel rappresentazione tradizionale (non delocalizzata) di doppi legami coniugati (–X=Y–Z=W–) 2 doppi legami π si trovano separati da un legame semplice σ. Gli orbitali p si sovrappongono 2 a 2 ed è sufficiente un’unica formula di struttura per descrivere la molecola

In realtà il sistema coniugato presenta i 4 orbitali p tra loro completamente sovrapposti e gli elettroni π risultano pertanto delocalizzati su 4 atomi.

Per rappresentare la delocalizzazione si utilizzano più formule di struttura, in cui il doppio legame si trova anche in posizione centrale. Il sistema coniugato π-π viene rappresentato con le seguenti tre strutture di risonanza

Un importante esempio di delocalizzazione elettronica in un sistema coniugato π-π si ha nel benzene C6H6. un composto organico in cui i 6 atomi di carbonio si chiudono a formare un esagono. Ciascun atomo di carbonio è ibridato sp2 ed impegna i tre orbitali sp2 per legarsi ad un idrogeno e ad altri due atomi di carbonio. L'orbitale pz non ibridato viene utilizzato per formare un legame π con un carbonio adiacente. Si dovrebbe pertanto ritenere che i 6 atomi di carbonio siano uniti all'interno dell'anello da una serie di legami semplici alternati a legami doppi.

In realtà i sei legami C - C risultano essere perfettamente identici e a metà strada tra un legame semplice ed un legame doppio.
Descriviamo dunque il benzene come un ibrido di risonanza delle due seguenti strutture limite
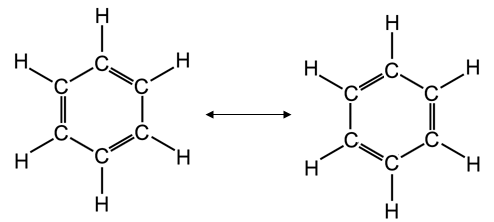
oppure, in modo del tutto equivalente, rappresentiamo le tre coppie di elettroni delocalizzati su tutta la molecola con un anello interno all'esagono

B) Sistema coniugato π-p
In un sistema coniugato π-p (–X=Y–Z*) l’orbitale p adiacente al doppio legame non è impegnato in alcun legame e può contenere da 0 a 2 elettroni (* = 0, 1, 2 elettroni)

Anche in questo caso il sistema coniugato presenta in realtà i 3 orbitali p tra loro completamente sovrapposti e gli elettroni risultano pertanto delocalizzati su 3 atomi.

Il sistema coniugato π-p viene rappresentato con due strutture di risonanza

A conferma di ciò i due legami non presentano la lunghezza tipica di un legame semplice ed uno doppio, ma hanno una lunghezza intermedia.
Si veda ad esempio la molecola dell’Ozono O3

I due legami O-O hanno una lunghezza di 128 nm, intermedia tra quella di un legame semplice O-O (149 nm) e quella di un legame doppio O=O (121 nm).
Nel caso l’orbitale p coniugato al doppio legame contenga una carica positiva o negativa, anche quest’ultima risulta delocalizzata.
Nel carbocatione allilico, ad esempio, la carica positiva è delocalizzata essendo portata per metà in C1 e per metà in C3

Nell’anione carbonato CO32- le due cariche negative sono distribuite su tutti e tre gli atomi di ossigeno

La mappa di potenziale elettrostatico dell’anione carbonato ci conferma la distribuzione di carica.

Regole di risonanza
- La posizione relativa degli atomi (connettività) in ciascuna struttura di Lewis deve rimanere la stessa. Può variare solo la posizione degli elettroni π e degli elettroni non condivisi (doppietti solitari ed eletroni spaiati o singoletti).
- Le strutture-limite devono essere valide strutture di Lewis. L’idrogeno non può condividere più di 2 elettroni . Gli elementi del secondo periodo non possono presentare più di 8 elettroni, sommando quelli condivisi (elettroni di legame) e quelli non condivisi (doppietti solitari). Gli elementi dei periodi superiori al secondo possono superare l’ottetto (ottetto espanso). Tra questi i più comuni sono il Fosforo (10 elettroni), lo Zolfo (12 elettroni) ed il Cloro (14 elettroni)..
- Ciascuna formula limite deve presentare il medesimo numero di elettroni totali, lo stesso numero di elettroni spaiati (se presenti) e la medesima carica netta.
- La risonanza si può verificare solo quando gli atomi coinvolti giacciono sullo stesso piano (o quasi); ogni variazione nella struttura che sia di ostacolo alla complanarità dei nuclei, impedisce o limita la risonanza (inibizione sterica della risonanza). Ciò è dovuto al fatto che la risonanza è un fenomeno di delocalizzazione elettronica.
Criteri di stabilità delle strutture risonanti
Abbiamo visto che le strutture-limite più stabili contribuiscono maggiormente all’ibrido. In altre parole l’ibrido assomiglia di più alle sue strutture-limite più stabili. Per valutare la stabilità relativa delle diverse strutture di risonanza si applicano, in ordine di importanza, i seguenti 4 criteri
- Sono più stabili le strutture limite che presentano il maggior numero di legami e quindi con il maggior numero di atomi che completano l’ottetto.

- A parità di legami sono più stabili le strutture-limite con il minor numero di cariche formali.

- A parità di cariche formali è più stabile la struttura-limite che presenta le cariche formali sugli elementi che meglio le “sopportano”. In questo caso si valutano principalmente l’elettronegatività, le dimensioni e l’ibridazione degli atomi che portano le cariche formali. Se gli atomi non presentano forti differenze nelle loro dimensioni (elementi appartenenti al medesimo periodo) si valutano le differenze di elettronegatività. Le strutture più stabili sono quelle in cui la carica negativa si trova sull’atomo più elettronegativo (o la carica positiva sull’atomo meno elettronegativo). La struttura più stabile per l’anione cianato è quella di sinistra poiché la carica negativa è portata dall’ossigeno (più elettronegativo dell’azoto).

La dimensione degli atomi va generalmente valutata per atomi che appartengano al medesimo gruppo chimico, aventi quindi la medesima configurazione elettronica superficiale, ma diversa dimensione (il raggio atomico aumenta scendendo lungo un gruppo). Le cariche negative sono meglio “sopportate” da atomi più grandi, che riescono in tal modo a disperderle su di un maggior volume atomico diminuendo la densità di carica. Le cariche positive sono meglio “sopportate” da atomi più piccoli, in cui gli elettroni di valenza, trovandosi più vicini al loro nucleo, risultano più saldamente legati.
La struttura più stabile per l’anione S-metantioato è quella di sinistra poiché la carica negativa è portata dallo Zolfo che, pur essendo meno elettronegativo dell’Ossigeno, presenta dimensioni atomiche maggiori.

Infine le cariche negative sono meglio sopportate da atomi in cui l’ibridazione ha un maggior carattere s. Un’orbitale sp, avendo un 50 % di carattere s, tiene gli elettroni più vicini al nucleo di un orbitale sp2 (33% di carattere s) o di un orbitale sp3 (25% di carattere s). Nell’esempio seguente la struttura più stabile è quella di destra

- Nel caso in cui due strutture presentino separazione di carica e le cariche siano localizzate sugli stessi tipi di atomi. la struttura più stabile è quella che presenta le cariche più vicine (minor separazione di carica). Nell’esempio seguente la struttura più stabile è quella di sinistra (senza cariche formali), ma tra le rimanenti due strutture la meno stabile è quella di destra che presenta la maggior separazione di carica.

- Da evitare. Alcune configurazioni, anche se possibili, risultano talmente instabili che il loro contributo all’ibrido di risonanza risulterebbe del tutto trascurabile. Per questo motivo è opportuno evitare di prenderle in considerazione. In particolare si evitino strutture con:
- cariche dello stesso segno su atomi adiacenti
- più di due cariche formali in più rispetto alla carica del composto
- più di una carica formale su di un medesimo atomo
- N e O con 6 elettroni (N ed O devono sempre completare l’ottetto, a differenza del C per il quale si accettano configurazioni con 6 elettroni)
- più di due legami in meno rispetto alla struttura più stabile
fonte: http://www.pianetachimica.it/didattica/documenti/Chimica_Generale.doc
Chimica generale 4
Visita la nostra pagina principale
Chimica generale 4
Termini d' uso e privacy