
Chimica generale 6
Chimica generale 6
Reticolo Esagonale Compatto – EC (hcp - Hexagonal closest packing)
12 atomi sono disposti ai vertici di un prisma a base esagonale, 2 atomi al centro di ciascuna base e 3 atomi all’interno della cella, a metà altezza, a formare i vertici di un triangolo equilatero

Ciascun atomo è circondato da altri 12 atomi

Le sfere del secondo strato giacciono nelle depressioni create dal primo strato, mentre le sfere del terzo strato giacciono nelle depressioni create dal secondo strato in modo da trovarsi esattamente sopra le sfere del primo strato

Ne deriva una successione di strati esagonali di tipo ABA

Il reticolo esagonale compatto è caratterizzato da un cella elementare prismatica esagonale che contiene 12 atomi posizionati ai vertici, 2 atomi posizionati al centro delle basi e 3 atomi ai vertici di un triangolo equilatero posizionati a metà altezza all’interno della cella elementare.
I 3 atomi interni appartengono completamente alla cella elementare. Ciascuno dei 12 atomi ai vertici è condiviso con altre 5 celle e quindi conta per 1/6 (contenuto in atomi = 12 x 1/6 = 2), mentre ciascuno dei due atomi posizionati al centro delle basi è condiviso con un'altra cella e conta per ½ (2 x 1/2 = 1) Quindi in totale vi sono 6 atomi equivalenti per cella.
Il lato dell’esagono di base costituisce la costante reticolare a ed essendo costituito da due sfere a contatto vale
a = 2r

Calcoliamo ora il volume del prisma esagonale in funzione del raggio atomico r.
Consideriamo il sito tetraedrico costituito da 2 sfere poste su due vertici della base, dalla sfera posta al centro della base e da una sfera posta all’interno del tetraedro.

Il tetraedro ha un’altezza h/2 pari alla metà dell’altezza h del prisma ed è formato da 4 facce equilatere di lato a. L’altezza b di una faccia (triangolo equilatero) del tetraedro coincide con l’apotema dell’esagono di base del prisma e vale
b = (a · Ö3) / 2
L’altezza del tetraedro è
h/2 = (a · Ö6) / 3
e quindi l’altezza del prisma vale
h = (2a · Ö6) / 3
L’area di base A del prisma è

Il volume V del prisma

e, poiché a = 2r, il volume V, espresso in funzione del raggio atomico r, diventa

Il coefficiente di impaccamento (fattore di compattazione atomica) si ottiene come rapporto tra il volume degli atomi equivalenti (sfere contenute nella cella elementare, in questo caso 6) ed il volume della cella elementare.

Reticolo |
|
Esempi |
Ti, Zn, Mg, Be |
Numero di atomi per cella elementare |
6 (cella a base esagonale) |
Numero di coordinazione |
12 |
Distanza tra atomi più vicini |
a |
Coefficiente di impaccamento |
Ö2 · p / 6 = 0,74 = 74% |
Esempio
Sapendo che il Titanio (d = 4,507 g/cm3 – PM = 47,867 g/mol) presenta una struttura esagonale compatta, calcoliamo il suo raggio atomico
Il volume della cella elementare può essere calcolato facendo il rapporto tra la massa di 6 atomi di Titanio (presenti nella sua cella elementare) e la densità del Titanio.
Calcoliamo la massa di 6 atomi di Titanio

Calcoliamo il Volume della cella elementare

Ricordando che il volume di una cella esagonale compatta in funzione del raggio atomico vale

possiamo ora calcolare il raggio atomico

Raggio metallico
Il raggio metallico viene spesso definito come la metà della distanza che separa i nuclei di atomi adiacenti nel solido a temperatura e a pressione ambiente.
Tuttavia tale distanza dipende dal numero di coordinazione e generalmente, cresce con esso, come si osserva studiando i polimorfi dei metalli (metalli che cristallizzano con diverse celle elementari) e i composti intermetallici (Goldschmidt)
Per confrontarei vari elementi si corregge la distanza ínternucleare empirica riportandola al valore prevedibile per l'elemento in un ipotetico impacchettamento compatto (numero di coordinazione. = 12) dividendo il raggio osservato per i seguenti fattori di correzione
Numero Coordinazione |
Fattore di correzione |
12 |
1 |
8 |
0,97 |
6 |
0,96 |
4 |
0,88 |
Ad esempio il raggio metallico del Sodio con N.C = 8 è 1.85 Å. Dividendo tale valore per 0,97 si ottiene 1,91 Å, il raggio che il Sodio avrebbe se fosse compatto.
I valori «corretti» di Goldschmidt sono quelli normalmente tabulati.
Modello a bande
Il modello di Drude è oggi sostituito da un modello quantistico del legame metallico che si deve a F.Bloch, ed e' conosciuto come modello a bande.
Il modello a bande è un’applicazione della Teoria dell’Orbitale Molecolare (MO) ai metalli.
Si consideri ad esempio la molecola Li2. Ciascun atomo di Litio presenta un orbitale atomico 2s semisaturo. La molecola forma pertanto due orbitali molecolari sigma: un orbitale legante (σ2s) saturo ed un orbitale antilegante (σ2s*) vuoto.

Si considerino ora 4 atomi di Litio legati. In questo caso si formeranno 2 orbitali di legame (σ2s) saturi e due orbitali di antilegame (σ2s*) vuoti. Questo sistema a 4 atomi è più stabile del precedente essendo caratterizzato da un maggior numero di orbitali di legame saturi.
 .
.
Se estendiamo il ragionamento ad n atomi otterremo un sistema costituito da n/2 orbitali di legame (σ2s) saturi ed n/2 orbitali di antilegame (σ2s*) vuoti. Gli orbitali di legame saturi e quelli di antilegame vuoti si dispongono su livelli energetici talmente vicini da formare una banda continua di energia.

In altre parole all'interno di ciascuna banda le differenza energetiche tra gli orbitali molecolari sono così piccole che possiamo considerare la distribuzione energetica come non quantizzata.

Come per il legame covalente, anche nel caso del legame metallico la forza del legame dipende dalla differenza tra il numero di elettroni negli orbitali leganti ed il numero di elettroni negli orbitali antileganti. Quanti più elettroni vi sono negli orbitali di legame rispetto agli orbitali di antilegame e tanto più il legame sarà intenso ed il metallo presenterà maggiore durezza e più elevato punto di fusione.
Questo significa che queste proprietà saranno massime al centro di un periodo di transizione. Per metalli con configurazione del tipo nd5 o adiacenti. In questo caso infatti, poiché ogni atomo ha i propri orbitali esterni semisaturi, gli orbitali molecolari leganti saranno completi, mentre quelli antileganti saranno vuoti. È una situazione analoga a quella del Litio, con la differenza che il numero di orbitali interessato al legame è notevolmente superiore.
Durezza e punto di fusione dei metalli hanno quindi un andamento periodico. Crescono dall’inizio fino al centro di una serie di transizione per poi decrescere proseguendo verso destra.

In un metallo la banda più esterna che contiene elettroni è detta banda di valenza Le bande possono essere separate da brevi intervalli energetici, dette zone proibite (band gap), in cui gli elettroni non possono essere presenti.
Il livello energetico più elevato occupato da elettroni (HOMO = Highest occupied molecular orbital) all’interno della banda di valenza alla temperatura dello zero assoluto è detto livello di Fermi (o energia di Fermi). Ad esempio per il Litio metallico il livello di Fermi si situa esattamente a metà della banda di valenza 2s.
Dunque a zero gradi kelvin gli elettroni del Litio, si trovano tutti ad energie minori dell’energia di Fermi, che si colloca a metà della banda 2s.

Ma a temperature superiori a zero kelvin gli elettroni, in quanto fermioni, obbedediscono alla distribuzione statistica di Fermi-Dirac. Anche per piccoli aumenti di temperatura, si rendono disponibili (nel senso statistico del termine) stati conduttivi, ossia elettroni che prima erano “congelati” nella metà inferiore della banda 2s, possono essere promossi ad energie più elevate e dunque risultano liberi di muoversi nel metallo e di condurre energia.

In un tale conduttore esistono stati liberi al di sopra del livello di Fermi e se si applica un campo elettrico gli elettroni si muovono nella direzione opposta al campo contribuendo a creare corrente. Al processo di conduzione non partecipano le bande di energia inferiore che sono totalmente occupate. Le bande ad energia inferiore, costituite dagli orbitali più interni e completamente sature, sono in genere poco estese e ben distanziate energeticamente (intervalli proibiti estesi). All’aumentare dell’energia le bande diventano più larghe e più ravvicinate fino, in alcuni casi, a sovrapporsi.
Nei metalli che, come il Litio, presentano la banda di valenza non completamente piena, banda di valenza e banda di conduzione coincidono.
In altre parole si tratta di considerare che, se un metallo presenta la banda di valenza non completamente piena di elettroni, essa è disponibile ad essere popolata da elettroni con energia sufficiente e diventare così una banda di conduzione. Questo meccanismo avviene tipicamente ad opera di schemi di eccitazione termica. Se una banda non è completamente piena, gli elettroni ad energia più alta possono facilmente essere promossi a stati di energia leggermente superiore, e contribuiscono significativamente alla conducibilità.
Questo accade per il Litio e per tutti i metalli che presentano una banda di valenza semisatura, ma è possibile anche per un metallo come il Berillio che, pur avendo la banda 2s completamente satura (sia quella di legame che quella di antilegame), presenta la banda 2s parzialmente sovrapposta alla banda 2p vuota che diventa dunque la banda di conduzione

Presentano dunque un comportamento metallico gli elementi con:
- la banda di valenza occupata solo parzialmente che funge da banda di conduzione
- la banda di valenza satura parzialmente sovrapposta alla banda di conduzione vuota
La conducibilità dei metalli diminuisce all'aumentare della temperatura poiché l'aumento dei moti vibrazionali dagli atomi va ad interferire con il moto degli elettroni.
La facilità con cui gli elettroni di conduzione possono muoversi attraverso il reticolo metallico spiega anche la buona conducibilità termica dei metalli. Quando un metallo viene avvicinato ad una fonte di calore gli elettroni di conduzione aumentano la loro energie cinetica media che, data la loro mobilità può essere facilmente trasferita alle particelle adiacenti.
La lucentezza dei metalli si spiega infine con la vicinanza degli orbitali molecolari all'interno della banda di conduzione. In pratica gli elettroni, avendo a disposizione moltissimi livelli energetici adiacenti, possono facilmente esservi promossi assorbendo luce su tutte le lunghezze d'onda per poi riemetterla per tornare allo stato fondamentale.
La teoria delle bande, oltre a giustificare le caratteristiche metalliche è in grado di fornire una spiegazione semplice ed immediata dell'esistenza dei semiconduttori e degli isolanti.
Isolanti
Gli isolanti sono caratterizzati da un sistema di bande nel quale quella più alta occupata è completamente piena (banda di valenza) e quella successiva, completamente vuota (banda di conduzione), si trova separata da un intervallo proibito talmente esteso da non poter essere superato se non sottoponendo il materiale a differenze di potenziale estremamente elevate.
La caratteristica di un isolante è quindi quella di possedere una banda di valenza completamente satura separata dalla banda di conduzione vuota da un intervallo di energia proibito molto maggiore della tipica energia termica. In tali condizioni la temperatura non è in grado di promuovere un elettrone nella banda superiore che è vuota e dunque, in presenza di un campo elettrico, non si ha passaggio di coerente. Un tipico isolante è il diamante in cui l'intervallo di energia proibito è circa duecento volte l'energia termica a temperatura ambiente. Alcuni fisici separano gli isolanti dai semiconduttori ponendo arbitrariamente pari a 4 eV le dimensioni energetiche della zona proibita.

semiconduttori
Sono semiconduttori elementi come il silicio ed il germanio che presentano una banda piena ed un intervallo di banda (zona proibita) con un valore non eccessivamente alto, tale comunque da poter essere superato fornendo adeguate quantità di energia al cristallo.
E' questo il motivo per cui nei semiconduttori la resistenza al passaggio di corrente elettrica diminuisce all'aumentare della temperatura. Le proprietà di semiconduttori o di isolanti dipendono quindi dall’intevallo (gap) tra la banda di valenza e quella di conduzione.

I semiconduttori hanno bassa conducibilità elettrica a temperatura ambiente, ma questa aumenta fortemente all’aumentare della temperatura. Il livello di Fermi si posiziona tipicamente tra la banda di valenza e quella di conduzione. All’aumentare della temperatura la distribuzione di Fermi-Dirac si modifica aumentando la probabilità che gli elettroni possiedano energia sufficiente a popolare la banda di conduzione.

Semiconduttori con particolari caratteristiche si possono costruire attraverso il processo di drogatura, aggiungendo ad un semiconduttore piccole percentuali di impurezze. Ad esempio mescolando al silicio piccole, ma ben definite quantità di arsenico o di gallio.
La drogatura con arsenico è detta di tipo n (negativa) in quanto viene aggiunto un elemento chimico che presenta la stessa configurazione superficiale del silicio più un elettrone. Gli elettroni in più vanno a disporsi nella banda superiore e sono disponibili per la conduzione.
La drogatura con gallio viene detta di tipo p (positiva) in quanto viene aggiunto un elemento chimico che presenta la stessa configurazione superficiale del silicio meno un elettrone. Gli elettroni in meno creano delle lacune elettroniche nella banda più superficiale del silicio creando le premesse per la conduzione.
Legami intermolecolari e forze di van der Waals
L'esistenza di aggregati di materia allo stato solido e liquido ci induce a ritenere che esistano delle forze anche tra molecole neutre in grado di legarle. Tali forze si producono sia tra molecole polari che tra molecole apolari e sono conosciute come forze di van der Waals. Le forze o interazioni di van der Waals, note anche come interazioni di non legame o legami deboli (definizione ambigua poichè in fisica le interazioni deboli sono una delle quattro forze fondamentali di natura), hanno un’intensità (0.1 - 10 kJ mol–1) che è mediamente di circa due ordini di grandezza inferiore all’intensità di un legame covalente o ionico (100 - 1000 kJ mol–1). Inoltre tali forze hanno un raggio d’azione estremamente breve, indebolendosi rapidamente all’aumentare della distanza.
L’energia di tali legami è infatti inversamente proporzionale alla sesta potenza della distanza che separa le particelle interagenti (E µ 1/r6), mentre le forze di van der Waals decrescono secondo la settima potenza della distanza (FvdW µ 1/r7).
Esistono tre tipi di forze di van der Waals:
- Forze di Keesom tra molecole polari
- Forze di Debye tra molecole polari e molecole apolari
- Forze di London tra molecole apolari
Forze di Keesom o interazioni dipolo-dipolo (effetto di orientazione)
Le molecole polari, o dipoli permanenti (molecole dotate di un momento di dipolo m), esercitano naturalmente una reciproca attrazione elettrostatica. Quando le molecole dipolari si avvicinano tendono infatti a disporsi con i poli di carica opposta l'uno di fronte all'altro, al fine di rendere minima l'energia potenziale del sistema (configurazione di maggior stabilità). In tal modo si verifica un'attrazione elettrostatica tra i poli opposti, detta interazione dipolo-dipolo. Le forze di Keesom agiscono dunque tramite un effetto di orientazione.
Le interazioni dipolo-dipolo non sono molto efficienti finché le molecole si trovano allo stato aeriforme poiché le distanze intermolecolari sono troppo elevate. Finché la temperatura è sufficientemente elevata e/o la pressione bassa, l'energia cinetica media dei dipoli è in grado di vincere tali interazioni, mantenendo la sostanza allo stato aeriforme. Ma all'abbassarsi della temperatura e/o all’aumentare della pressione, le distanze intermolecolari diminuiscono e l'energia cinetica media delle molecole finisce per diventare minore delle interazioni dipolari. In queste condizioni tali forze sono in grado di mantenere adese le molecole favorendo il passaggio ad una fase condensata (liquida o solida). Inizialmente si ha il passaggio allo stato liquido e, se la temperatura scende ulteriormente (o la pressione aumenta), le forze di Keesom sono in grado di bloccare le molecole in posizioni di equilibrio all'interno di un reticolato solido.

Interazioni dipolo-dipolo tra molecole polari allo stato solido
Le interazioni dipolo-dipolo sono ovviamente tanto più intense quanto maggiore è il momento di dipolo m ed iniziano a diventare importanti per valori di m superiori ad 1 D. La loro intensità decresce all’aumentare della temperatura, poiché una maggior agitazione termica interferisce con l’allineamento dei dipoli.
Per due dipoli liberi di ruotare (liquido o aeriforme) di momento m1 ed m2 a distanza r l’energia di Keesom è

con
k = costante di Boltzmann = 1,38 10-23 J K-1
εo = costante delettrica del vuoto = 8,854 10-12 C2 m-2 N-1
T = temperatura assoluta
Ad esempio, a 25°C l’energia di interazione per una coppia di molecole con μ = 1 D (= 3,336 10-30 C m) alla distanza di 0.3 nm (= 3 10-10 m) è di

L’energia di Keesom per una mole si ottiene moltiplicando il risultato precedente per il numero di Avogadro N = 6.022 1023 mol-1.
- 2,22 10-22 x 6.022 1023 = - 1340 J mol-1 = - 1,34 kJ mol-1
Per due dipoli stazionari (all’interno di un solido) l’energia di Keesom risulta inversamente proporzionale alla terza potenza della distanza r e dipende dall’orientazione reciproca (angoli θ e φ).


Quando il dipolo è costituito da un atomo di idrogeno legato con legame covalente fortemente polare ad un elemento molto elettronegativo (F, O, N), il legame dipolo-dipolo è particolarmente intenso e viene chiamato legame a idrogeno (o ponte idrogeno). I legami a idrogeno presentano energie tipiche superiori (20 – 50 kJmol-1) rispetto ai normali legami dipolo-dipolo.
Il legame a idrogeno viene rappresentato con una breve linea tratteggiata che unisce l'idrogeno di una molecola con l'elemento elettronegativo di un'altra.
Tipici composti in grado di dare intensi legami a idrogeno sono l'acido fluoridrico HF, l'acqua H2O e l'ammoniaca NH3.

Legami a idrogeno tra molecole d’acqua e ammoniaca
L'esistenza di tale legame aumenta notevolmente la coesione interna tra le molecole, al punto da riflettersi in modo evidente su alcune proprietà fisiche delle sostanze interessate.
Ad esempio tutti i composti le cui molecole sono interessate dai legami a idrogeno presentano temperature di ebollizione e capacità termiche particolarmente elevate.
Se infatti forniamo calore ad una sostanza produciamo un aumento della sua energia cinetica media (½mv²). E' allora evidente che a parità di calore fornito l'aumento di velocità sarà minore per le molecole più massicce. Poiché inoltre una sostanza è in grado di passare allo stato di vapore quando le sue molecole sono sufficientemente veloci, dobbiamo attenderci che la temperatura di ebollizione di un composto sia tanto maggiore quanto maggiore è il suo peso molecolare.
Tale previsione è verificabile osservando ad esempio i composti dell'idrogeno con gli elementi del VII gruppo A, dove il punto di ebollizione diminuisce costantemente al diminuire del peso molecolare, con la notevole eccezione dell'acido fluoridrico.

In questo caso infatti, nonostante il basso peso molecolare, la temperatura di ebollizione risulta particolarmente elevata in quanto per poter passare allo stato di vapore le molecole devono possedere un'energia cinetica molto elevata per rompere i legami a idrogeno che le tengono adese.
La presenza del legame a idrogeno spiega anche perchè il ghiaccio sia meno denso dell'acqua. Infatti quando l'acqua si solidifica i legami a idrogeno tendono a bloccare le molecole in una struttura esagonale ordinata che risulta meno densa della struttura disordinata caratteristica dell'acqua liquida.

Forze di Debye o interazioni dipolo permanente-dipolo indotto (effetto di induzione)
Le forze di Debye si originano tra molecole polari e molecole apolari. Per comprendere tali interazioni è necessario esaminare ciò che accade ad una molecola (o un atomo) apolare quando viene posta in un campo elettrico E.

a) molecola non polarizzata b) Polarizzazione in un campo elettrico E
La nuvola elettronica della molecola viene deformata ed attratta dal polo positivo. Il campo elettrico induce dunque una separazione di carica con formazione di un dipolo indotto. L’intensità del momento di dipolo indotto m è direttamente proporzionale all’intensità E del campo elettrico applicato.
m = a E
La costante di proporzionalità a è detta polarizzabilità. Il valore della polarizzabilità è caratteristico per ciascun atomo o molecola ed è una misura della facilità con cui la nuvola elettronica può essere deformata (polarizzata) da un campo elettrico.
L'unità di misura della polarizzabilità nel Sistema Internazionale è C·m2·V-1.
Spesso invece della polarizzabilità si usa il volume di polarizzabilità α', definito da:

dove εo è la costante dielettrica del vuoto.
α' ha le dimensioni di un volume. L'unità di misura utilizzata nella pratica comune per esprimere il suo valore è il cm3 o Å3.
La polarizzabilità dipende dalla forza con cui gli elettroni esterni sono vincolati al nucleo (minore è l’energia di ionizzazione, maggiore è la polarizzabilità). In altre parole, la nuvola elettronica è tanto più facilmente polarizzabile quanto minore è la forza di attrazione che il nucleo esercita su di essa. La polarizzabilità aumenta all’aumentare della massa e delle dimensioni della molecola.
- All’aumentare della massa aumenta infatti il numero di elettroni. La carica nucleare risulta pertanto maggiormente schermata dai gusci elettronici più interni. Questo effetto di schermatura permette agli elettroni superficiali di “sentire” in misura minore la carica del loro nucleo e di risultare quindi meno legati.
- All’aumentare del volume aumenta la distanza degli elettroni più esterni dal nucleo e diminuisce di conseguenza la forza attrattiva su di essi esercitata dal nucleo.
Quando una molecola polare si avvicina ad una non polare induce in quest'ultima un dipolo eletrico di minore intensità (effetto di induzione) che perdura fintanto che le due molecole restano vicine. Si genera così un’attrazione dipolo permanente-dipolo indotto . L'intensità è proporzionale al momento del dipolo permanente m che induce la polarizzazione e alla polarizzabilità a della seconda molecola.

catione
Per un dipolo di momento m ed una molecola apolare di polarizzazione a a distanza r l’energia di Debye è

con εo = costante delettrica del vuoto = 8,854 10-12 C2 m-2 N-1
Ad esempio, a 25°C l’energia di interazione per un dipolo (ad esempio HCl) con μ = 1 D (= 3,336 10-30 C m) ed una molecola apolare (ad esempio il benzene) con polarizzazione a’ = 10-23 cm3 (= 10-29 m3) alla distanza di 0.3 nm (= 3 10-10 m) è di

L’energia di Debye per una mole si ottiene moltiplicando il risultato precedente per il numero di Avogadro N= 6.022 1023 mol-1.
- 1,37 10-21 x 6.022 1023 = - 820 J mol-1 = - 0,82 kJ mol-1
Forze di London o interazioni dipolo istantaneo-dipolo indotto (effetto di dispersione)
Se anche le molecole perfettamente apolari come O2 e Cl2 sono in grado di liquefare e solidificare a temperature superiori allo zero assoluto, evidentemente devono esistere anche per tali sostanze delle forze intermolecolari, seppur molto deboli, in grado di vincere l'agitazione termica.
Si ritiene che tali forze, dette forze di London o forze di dispersione, siano dovute a fluttuazioni temporanee e casuali nella distribuzione di densità degli orbitali. In una molecola apolare la nuvola elettronica è “in media” distribuita in modo omogeneo, ma in un determinato istante questo può non essere vero e gli elettroni possono casualmente e temporaneamente essere addensati a formare un dipolo istantaneo (o dipolo momentaneo o dipolo temporaneo).

Piccole fluttuazioni nella distribuzione delle nuvole elettroniche dovrebbero essere dunque in grado di produrre momentanee polarità anche nelle molecole apolari capaci di indurre nelle molecole adiacenti polarità di segno contrario (dipolo indotto), creando in definitiva le condizioni per un'attrazione reciproca.
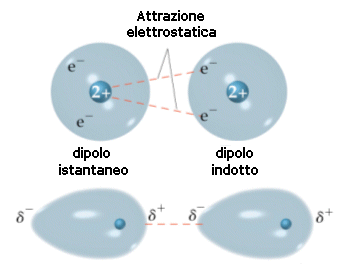
Se si considera la media nel tempo, la nuvola elettronica di un atomo è perfettamente simmetrica, ma in un dato istante può addensarsi maggiormente da un lato ed in un istante immediatamente successivo può spostarsi all’altra estremità. Ciò determina la comparsa di un momento di dipolo elettrico istantaneo variabile nel tempo e mediamente nullo. Ciascun dipolo istantaneo genera un campo elettrico che polarizza le particelle circostanti, creando dei “dipoli indotti” variabili continuamente. Tra il dipolo “induttore” e il dipolo “indotto” nascono così forze di attrazione.

L’intensità delle forze di London dipende ovviamente solo dalla polarizzabilità a delle molecole. Molecole più grandi e massicce (con elettroni superficiali meno legati) risentono in misura maggiore delle forze di London. Ad esempio, a temperatura ambiente, mentre F2 e Cl2 sono gassosi, Br2 è liquido e I2 è solido.

Per due molecole apolari aventi polarizzazione a1 ed a2 a distanza r l’energia di London è

dove
I = hn è l’energia di ionizzazione
con
h = costante di Planck = 6.626 10-34 J s
n = frequenza principale di assorbimento della molecola
Per due molecole identiche la relazione diventa

Nel caso di due molecole di metano, ad esempio, con α' = 2.6·10-30 m3 e I = 7 eV (= 1,12·10-18 J) l’energia di dispersione alla distanza di 0.3 nm (= 3·10-10 m) è di

L’energia di London per una mole si ottiene moltiplicando il risultato precedente per il numero di Avogadro N= 6.022 1023 mol-1.
- 7,79·10-21 x 6.022·1023 = - 4690 J mol-1 = - 4,69 kJ mol-1
Le forze di London sono universali, essendo presenti anche in tutti i tipi di atomi e molecole e, nella maggior parte dei casi, costituiscono la componente prevalente delle forze di van der Waals anche in molecole polari. Affinché le interazioni dipolo-dipolo (forze di Keesom) inizino ad essere significative prevalenti rispetto alle forze di London è infatti necessario che le molecole siano di piccole dimensioni (poco polarizzabili) ed abbiano un momento di dipolo superiore ad 1 D (esempi tipici sono l’acqua e l’ammoniaca). Le forze di induzione (forze di Debye) risultano invece per lo più trascurabili.
|
μ |
α' |
En Ionizz. |
Keesom |
Debye |
London |
Ar |
0 |
1.63 |
15.4 |
0 |
0 |
4 |
CO |
0.40 |
1.99 |
14.3 |
2,4·10-3 |
2,4·10-3 |
5,62 |
HCl |
3.50 |
2.63 |
13.7 |
1,5 |
0,23 |
9,41 |
HBr |
2.67 |
3.61 |
13.3 |
0,49 |
0,18 |
17,2 |
HI |
1.40 |
5.44 |
12 |
2,8·10-2 |
6,5·10-2 |
35,2 |
NH3 |
4.87 |
2.26 |
16 |
6,6 |
0,4 |
8,11 |
H2O |
6.17 |
1.59 |
18 |
15,7 |
0,42 |
4,52 |
Valori calcolati a 25°C per una distanza intermolecolare di 0,3 nm (3 Å) |
||||||
Repulsione di van der Waals a corto raggio e potenziale di Lennard-Jones
In generale, l’interazione attrattiva totale fra molecole neutre, polari o apolari, denominata attrazione di van der Waals, dipende dal contributo delle interazioni descritte precedentemente, dipolare (di orientazione), di induzione e di dispersione. Dato che nella fase fluida tutte tre dipendono dall’inverso della sesta potenza della distanza si può esprimere unitariamente l’energia di attrazione come

dove la costante di proporzionalità A dipende dalla natura delle molecole interagenti.
Le forze di attrazione di van der Waals prevalgono alle distanze intermolecolari maggiori. Esse hanno un range compreso fra qualche Ǻ ed un centinaio di Ǻ.
A distanze inferiori di qualche Ǻ entrano tuttavia in gioco forze repulsive. L’effetto repulsivo a corto raggio, chiamata repulsione sterica o repulsione di van der Waals, si genera tra i nuclei che, a distanze piccole, non sono più ben schermati dagli elettroni, e fra gli elettroni stessi, soggetti a una forza repulsiva che si genera quando due o più di essi tendono ad occupare gli stessi numeri quantici, in opposizione al principio di Pauli.
Tali forze repulsive, caratterizzate da un raggio d'azione molto breve, crescono rapidamente all'avvicinarsi delle molecole. Il calcolo delle interazioni tra coppie di molecole a brevi distanze presenta notevoli difficoltà. Per esse non esiste infatti un'equazione ricavata teoricamente che le descriva e ci si affida quindi ad alcune funzioni potenziali empiriche. E’ richiesto solo che esse tendano a zero per r che tende all’infinito più velocemente del termine r6.
Una drastica approssimazione, che in qualche caso si adotta per semplificare la trattazione, consiste nell'assumere per le repulsioni a corto raggio la forma
E = 0 per r > s
E = ∞ per r ≤ s
note come repulsioni a sfera rigida (hard-sphere).

dove σ, detto diametro di collisione, è il diametro della sfera che approssima l'atomo o la molecola. È la distanza di massimo avvicinamento di due molecole e dunque la distanza che separa i due nuclei quando le molecole urtano ed al di sotto della quale la repulsione diventa infinita (impenetrabilità).

Per determinare s si utilizza spesso la somma dei raggi di van der Waals delle molecole interagenti.
Una approssimazione migliore e molto utilizzata rispetto al modello a sfera rigida è quella di Lennard-Jones in cui la parte repulsiva ha una dipendenza da r12

dove la costante di proporzionalità B dipende, ancora una volta, dalla natura delle molecole. La repulsione cresce inversamente alla distanza r fra gli atomi elevata alla dodicesima potenza, cioè molto bruscamente. Non esistono argomenti teorici in favore dell’esponente 12, che è stato scelto solo per convenienza di calcolo (essendo il quadrato del termine r6).
Per ottenere l’energia netta dell’interazione intermolecolare si sommano le energie di repulsione e di attrazione viste precedentemente

Questa relazione si scrive solitamente nella forma del potenziale di Lennard-Jones

in cui compaiono i parametri σ e ε, il cui valore dipende dal tipo di atomi.
Il primo (σ) ha le dimensioni di una lunghezza ed è la distanza alla quale il potenziale si annulla.
Il secondo (ε) ha le dimensioni di un'energia e rappresenta la profondità della buca di potenziale e dunque l’energia di interazione intermolecolare.
I due parametri possono essere messi in relazione rispettivamente con il diametro atomico e con la massima energia di attrazione tra una coppia di molecole. Se A e B sono molecole diverse, si definisce
 .
.
Il potenziale di Lennard-Jones è particolarmente adatto per simulazioni di gas nobili

Interazioni tra ioni e molecole neutre
Un particolare tipo di interazioni intermolecolari possono essere considerate quelle che si manifestano tra ioni e molecole neutre, sia polari che apolari. L’intensità di tali interazioni è superiore a quella delle forze di van der Waals ((0.1 - 10 kJ mol–1), ma inferiore alle forze di legame (covalente, ionico e metallico) ed è dell’ordine di 101- 102 kJ mol–1.
- Interazione Ione-Dipolo permanente
Questa interazione è all'origine della solubilità delle sostanze ioniche in acqua. Durante il processo di idratazione (solvatazione) il catione attrae l’estremità negativa dei dipoli dell’acqua, l’anione l’estremità positiva. Il numero di molecole di acqua legate, denominato numero di idratazione dello ione, è direttamente proporzionale alla carica dello ione e inversamente proporzionale alla sua dimensione. Ioni piccoli come Li+, Na+, F-, OH- sono capaci di legare molecole di acqua nella prima sfera di idratazione e producono ordine anche oltre questa sfera, per questo si chiamano ioni strutturanti. Uno solido si scioglie se la sua energia reticolare è inferiore dell’energia di idratazione. I valori caratteristici di intensità di queste interazioni cadono nell’intervallo 40-600 kJ mol-1.

L'energia di interazione fra uno ione avente carica q e un dipolo permanente m, libero di ruotare (e quindi in fase fluida), ad una distanza r è

dove, al solito, il segno negativo indica che l'interazione è attrattiva, con
k = costante di Boltzmann = 1,38 10-23 J K-1
εo = costante delettrica del vuoto = 8,854 10-12 C2 m-2 N-1
T = temperatura assoluta
Ad esempio, a 25°C l’energia di interazione per uno ione monovalente (Q = 1,602 10-19 C) ed una molecola d’acqua con μ = 1,85 D (= 6,17 10-30 C m) alla distanza di 0.3 nm (= 3 10-10 m) è di

L’energia di interazione per una mole si ottiene moltiplicando il risultato precedente per il numero di Avogadro N = 6.022 1023 mol-1.
- 3,95 10-19 x 6.022 1023 = - 238000 J mol-1 = - 238 kJ mol-1
Per un dipolo stazionario l’energia di interazione con uno ione risulta inversamente proporzionale alla seconda potenza della distanza r e dipende dall’orientazione (angolo θ).


- Interazione Ione-Dipolo indotto
Il campo elettrostatico di uno ione è in grado di polarizzare un atomo o una molecola neutra ed apolare presente nelle vicinanze deformando la sua nuvola elettronica superficiale e generando un dipolo indotto, che subisce di conseguenza un’attrazione da parte della specie ionica.

L’intensità di questa interazione dipende dalla carica (Q) dello ione, dalla polarizzabilità ( ) della molecola (o dell’atomo) apolare e decresce con la quarta potenza della distanza (r).
) della molecola (o dell’atomo) apolare e decresce con la quarta potenza della distanza (r).

Ad esempio, a 25°C l’energia di interazione per uno ione monovalente (Q = 1,602 10-19 C) ed una molecola di tetraclorometano CCl4 con volume di poarizzabilità α' = 10.5·10-30 m3, alla distanza di 0.3 nm (= 3 10-10 m) è di

L’energia di interazione per una mole si ottiene moltiplicando il risultato precedente per il numero di Avogadro N = 6.022 1023 mol-1.
- 1,50 10-19 x 6.022 1023 = - 90100 J mol-1 = - 90,1 kJ mol-1
Si tenga presente che: un dipolo indotto si origina sempre nell'interazione tra uno ione ed una molecola (sia essa polare o no). Se la molecola è polare, la forza di attrazione dovuta all'interazione ione-dipolo indotto si somma a quella dovuta all'interazione ione-dipolo permanente.
In conclusione si riportano i diversi tipi di legami intermolecolari e si confrontano con i legami interatomici covalente e ionico:
Tipo di interazione |
F(r) |
Energia tipica |
Esempio |
|
Ione-specie neutra |
Ione-Dipolo permanente |
r-4 |
50-500 |
Na+ (H2O)n |
Ione-Dipolo indotto |
10-100 |
Na+ C6H6 |
||
tra specie neutre |
Dipolo perm-Dipolo perm |
r-6 |
0.5-15 |
SO2 SO2 |
Dipolo permanente-Dipolo indotto |
0.4-4 |
HCl C6H6 |
||
Dipolo istantaneo-Dipolo indotto |
4-40 |
CH4 CH4 |
||
Legame a idrogeno |
- |
4-40 |
H2O×××HOCH3 |
|
Ione-Ione |
Legame ionico |
r-1 |
40-400 |
Na+ Cl- |
Atomo-Atomo |
Legame covalente |
- |
200-800 |
H-H |
Costruzione dei composti e nomenclatura chimica
Per costruire correttamente la maggior parte dei composti chimici è sufficiente conoscere alcune semplici regole. Fondamentale è a questo proposito il concetto di numero di ossidazione di un elemento (nox) o stato di ossidazione (stox).
Numero di ossidazione (nox) o stato di ossidazione (stox)
Si definisce numero di ossidazione la carica, reale o formale, che acquista un atomo quando si assegnano convenzionalmente gli elettroni di legame all'atomo più elettronegativo.
La carica è reale nei composti ionici ed in tal caso coincide con il numero di cariche portate dallo ione.
Ad esempio nel cloruro di sodio NaCl, costituito da uno ione sodio Na+ e da uno ione cloro Cl-, il sodio presenta nox +1, mentre il cloro presenta nox -1.
La carica è formale nei composti covalenti. Ad esempio nell'acqua H2O, gli elettroni di legame vengono assegnati all'ossigeno più elettronegativo, il quale assume perciò convenzionalmente 2 cariche negative e presenta nox -2. Ciascuno dei due idrogeni presenta quindi nox +1.
Ciascun elemento chimico può presentare più di un numero di ossidazione. Vengono date di seguito alcune regole convenzionali per l'attribuzione dei numeri di ossidazione.
1) il nox delle sostanze elementari (H2, O2, Na, Cu etc) è sempre zero poiché ci troviamo di fronte ad atomi di uno stesso elemento, aventi perciò la stessa elettronegatività.
Più in generale quando in una molecola due atomi di uno stesso elemento si uniscono con legame covalente, gli elettroni di legame non vanno attribuiti a nessuno dei due atomi.
2) Il nox di uno ione è pari alla sua carica
Ca2+ (nox +2) Al3+ (nox +3) S2- (nox -2)
3) L'idrogeno presenta sempre nox +1 tranne che negli idruri, composti in cui si lega direttamente con i metalli alcalini e alcalino-terrosi che risultano essere gli unici elementi più elettropositivi dell'idrogeno. In tali composti l'idrogeno ha dunque nox -1.
Gli idruri si scrivono sempre facendo seguire al simbolo del metallo il simbolo dell'idrogeno.
idruro di sodio NaH, idruro di calcio CaH2 etc
4) L'ossigeno ha sempre nox -2 tranne che nei perossidi dove presenta nox -1. I perossidi sono composti dove l'ossigeno impegna uno dei suoi due elettroni per legarsi ad un altro atomo di ossigeno. secondo quanto previsto dalla regola numero 1 in questo caso gli elettroni del legame tra atomi uguali non vanno attribuiti, mentre viene attribuito all'ossigeno l'altro elettrone utilizzato per legarsi ad altri elementi
perossido di idrogeno o acqua ossigenata H2O2
H O O H
perossido di sodio Na2O2
Na O O Na
perossido di magnesio MgO2

5) il fluoro, essendo l'elemento più elettronegativo della tabella periodica, ed avendo bisogno di un solo elettrone per raggiungere l'ottetto, ha sempre nox -1
6) Gli altri elementi del VII gruppo A hanno anch'essi nox -1, tranne quando si legano con elementi più elettronegativi, come ad esempio l'ossigeno, in tal caso presentano nox positivi.
7) In generale il nox più elevato di un elemento corrisponde al numero d'ordine del gruppo cui appartiene. Così gli elementi del primo gruppo presentano nox +1, quelli del secondo +2, quelli del terzo +3 e così via fino agli elementi del settimo gruppi che presentano come nox più elevato +7.
8) sempre in generale, quando un elemento presenta più di un nox, il valore di quest'ultimo diminuisce di 2 unità alla volta.
Così gli elementi del VII gruppo oltre al nox +7 possono presentare nox +5, +3, +1, -1.
gli elementi del VI gruppo oltre al nox + 6 possono presentare nox +4, +2, -2.
9) In una specie chimica neutra la somma dei nox di tutti gli atomi che la compongono deve sempre essere nulla.
10) In uno ione poliatomico la somma dei nox dei diversi atomi deve sempre essere pari alla carica totale dello ione.
Le ultime due regole ci permettono, partendo da una formula chimica, di calcolare il numero di ossidazione incognito della maggior parte degli elementi.
Ad esempio per calcolare il numero di ossidazione dello zolfo nell'anidride solforica SO2, procediamo come segue: ciascun atomo di ossigeno presenta nox -2; complessivamente i due atomi presentano nox -4; affinché la somma dei nox sia zero lo zolfo deve presentare nox + 4.
Calcoliamo il nox del carbonio nello ione poliatomico HCO3-: i tre atomi di ossigeno presentano complessivamente nox - 6, l'idrogeno presenta nox + 1. Sommando il nox degli ossigeni e dell'idrogeno si ottiene - 5. Affinché la somma di tutti i nox dia la carica complessiva dello ione -1, il carbonio deve presentare nox +4.
La conoscenza dei numeri di ossidazione ci permette di costruire in modo semplice i principali composti chimici.
Regole per la costruzione dei composti binari
I composti binari sono formati da due soli elementi chimici.
Convenzionalmente si scrivono ponendo per primo l'elemento meno elettronegativo, seguito dall'elemento più elettronegativo.
Il simbolo di ciascun elemento è seguito da un numero a pedice, detto indice, che indica quanti atomi di quell'elemento sono presenti nel composto.
Gli indici sono apposti in modo tale che, sommando i rispettivi nox, la molecola risulti neutra.
Per calcolare gli indici in modo semplice è sufficiente utilizzare il nox del primo elemento come indice del secondo e viceversa.
Ad esempio se vogliamo scrivere la formula di un composto binario formato da un elemento A il cui numero di ossidazione sia +2 e da un composto B il cui numero di ossidazione sia -3, otterremo

Si noti che l'elemento con il numero di ossidazione negativo (il più elettronegativo) è stato scritto per secondo.
Tale metodo di costruzione dei composti binari garantisce la neutralità della molecola.
Infatti nella molecola sono presenti 3 atomi di A per un totale di 6 cariche positive e 2 atomi di B per un totale di 6 cariche negative.
Qualora dopo aver calcolato gli indici questi risultino divisibili per uno stesso numero, gli indici vanno semplificati, tranne alcuni casi particolari (vedi ad esempio alcuni perossidi).
Ad esempio se vogliamo costruire un composto binario partendo dagli elementi X con numero di ossidazione +4 e Y con numero di ossidazione -2, si otterrà

Fanno eccezione alcuni composti, la cui formula è necessario conoscere, come ad esempio il perossido di idrogeno, H2O2, in cui gli indici non vanno semplificati.
Principali composti binari
Idruri
Sono composti dell'idrogeno con metalli più elettropositivi. In tali composti l'idrogeno presenta nox -1 (ione idruro H-) e quindi nella formula va scritto per secondo.
Gli idruri dei metalli alcalini (I gruppo A) che presentano nox +1, hanno formula generale
MeH
Ad esempio idruro di potassio, KH
Gli idruri dei metalli alcalino terrosi (II gruppo A), che presentano tutti nox +2, hanno formula generale
MeH2
Ad esempio idruro di calcio, CaH2.
Il loro nome è formato dal termine "idruro" seguito dal nome del metallo
Perossidi
Sono composti in cui è presente il gruppo perossido ( O O ) unito ad elementi più elettropositivi. Nei perossidi ciascun atomo di ossigeno presenta nox -1.
Il loro nome è formato dalla parola "perossido" seguito dal nome dell'elemento legato.
Ad esempio
Perossido di idrogeno H2O2, perossido di bario BaO2.
Ossidi
Sono composti in cui un metallo si lega con l'ossigeno (nox -2).
Si formano per la reazione di un metallo con l'ossigeno
Metallo + O2 ® ossido
La reazione è rapida con i metalli dei primi gruppi, che presentano forte carattere metallico, più lenta con gli altri metalli.
Il loro nome è formato dalla parola "ossido" seguito dal nome del metallo.
I gruppo A |
Li2O, Na2O, K2O etc
II gruppo A |
BeO, MgO, CaO etc
III gruppo A |
il boro è un semimetallo Al2O3, Ga2O3 etc
IV gruppo A |
Gli unici metalli sono stagno e piombo che presentano nox +2 e +4, formando con l'ossigeno due tipi di ossidi. In tal caso il composto a nox maggiore prende la desinenza -ico, quello a nox minore prende la desinenza -oso.
Stagno (+2, +4) ossido stannoso SnO ossido stannico SnO2
Piombo (+2, +4) ossido piomboso PbO ossido piombico PbO2.
Il minio (antiruggine) Pb3O4, viene considerato un ossido salino o piombato piomboso (Pb2+)2PbO44-
Principali ossidi dei gruppi B
I gruppo B |
Rame (nox +1, +2) ossido rameoso Cu2O, ossido rameico CuO
Argento (+1) ossido d'argento Ag2O
Oro (+1,+3) ossido auroso Au2O, ossido aurico Au2O3
II gruppo B |
Zinco (+2) ossido di zinco ZnO
Cadmio (+2) ossido di cadmio CdO
VI gruppo B |
Mercurio (+1, +2) ossido mercuroso Hg2O ossido mercurico HgO
Il Cromo (+2, +3, +6) si comporta come un metallo con i numeri di ossidazione +2 e +3,
Cromo (+2, +3) ossido cromoso CrO ossido cromico Cr2O3
VII gruppo B |
Il Manganese (+2, +3, +4, +6, +7) si comporta come un metallo con i nox più bassi, mentre con il nox +4 forma il biossido di manganese MnO2 che presenta carattere anfotero
Manganese (+2, +3) ossido manganoso MnO ossido manganico Mn2O3
VIII gruppo B |
.
Ferro (+2, +3) ossido ferroso FeO ossido ferrico Fe2O3
Cobalto (+2, +3) ossido cobaltoso CoO ossido cobaltico Co2O3
Nichel (+2, +3) ossido nicheloso NiO ossido nichelico Ni2O3.
ANIDRIDI
Le anidridi sono composti binari dei non metalli con l'ossigeno.
Il loro nome è formato dalla parola "anidride" seguita dal nome del non metallo.
Non Metallo + O2 Anidride
VII gruppo A |
Cloro (+1, +3, +5, +7) anidride ipoclorosa (nox +1) Cl2O
anidride clorosa (nox +3) Cl2O3 (non è nota)
anidride clorica (nox +5) Cl2O5 (non è nota)
anidride perclorica (nox +7) Cl2O7
Con nox +4 forma il biossido di cloro ClO2
Bromo (+1, +5) anidride ipobromosa (nox +1) Br2O
anidride bromica (nox +5) Br2O5 (non è nota)
con nox +4 forma il biossido di bromo BrO2
Iodio (+1, +5, +7) anidride ipoiodosa (nox +1) I2O (non è nota)
anidride iodica (nox +5) I2O5
anidride periodica (nox +7) I2O7 (non è nota)
Il fluoro con nox -1 forma con l'ossigeno un composto estremamente instabile, l'ossido di fluoro F2O(dove l'ossigeno presenta nox +2). In realtà, essendo il fluoro più elettronegativo, andrebbe scritto OF2 e considerato un fluoruro di ossigeno.
VI gruppo A |
Zolfo (+4, +6) anidride solforosa (nox +4) SO2
anidride solforica (nox + 6) SO3
con i numeri di ossidazione +2 e +3 forma il protossido di zolfo SO e l'anidride iposolforosa (sesquiossido) S2O3.
Selenio (+4, +6) anidride seleniosa (nox +4) SeO2
anidride selenica (nox +6) SeO3
Tellurio (+4, +6) anidride tellurosa (nox +4) TeO2
anidride tellurica (nox +6) TeO3 (non è nota)
V gruppo A |
Azoto (+3, +5) anidride nitrosa (nox +3) N2O3
anidride nitrica (nox +5) N2O5
Con nox +4 forma l'ipoazotide N2O4, in equilibrio con il biossido di azoto NO2 (può essere considerato un'anidride mista nitroso-nitrica, infatti in acqua da una miscela di acido nitroso e nitrico)
con nox +2 forma il monossido di azoto NO (o ossido nitrico)
con nox +1 il protossido di azoto N2O (gas esilarante) o anidride iponitrosa (o ossido nitroso).
Fosforo (+3, +5) anidride fosforosa (nox +3) P2O3 (in realtà P4O6)
anidride fosforica (nox +5) P2O5 (in realtà P4O10)
con nox +4 forma il tetrossido di fosforo P2O4, analogo all'ipoazotide (in acqua da una miscela di acido fosforoso e fosforico)
Le anidridi degli altri tre elementi appartenenti al quinto gruppo (arsenico, antimonio e bismuto) possono essere anche classificati come ossidi dato il comportamento anfotero di tali composti.
Arsenico (+3, +5) anidride (o ossido) arseniosa ( nox +3) As2O3 (in realtà As4O6)
anidride (o ossido) arsenica ( nox +5) As2O5
con nox +4 forma il tetrossido di arsenico As2O4, analogo all'ipoazotide (può essere considerata un'anidride mista arsenioso-arsenica, infatti in acqua da una miscela dei due acidi corrispondenti)
Antimonio (+3, +5) anidride (o ossido) antimoniosa (nox +3) Sb2O3 (in realtà Sb4O6)
anidride (o ossido) antimonica (nox +5) Sb2O5
con nox +4 forma il tetrossido di antimonio Sb2O4, che si ritiene sia un ossido salino simile al minio o antimoniato antimonioso Sb3+(SbO4)3-.
Bismuto (+3, +5) ossido di bismuto (nox +3) Bi2O3
anidride (o ossido) bismutica (nox +5) Bi2O5
con nox +4 forma il tetrossido di antimonio Bi2O4.
IV gruppo A |
Carbonio (+2, +4) ossido di carbonio (nox +2) CO
anidride carbonica (nox +4) CO2
Silicio (+4) anidride silicica (o biossido) SiO2
Germanio (+4) Anidride germanica (o biossido) GeO2
III gruppo A |
Boro (+3) Anidride borica (o ossido) B2O3
VI gruppo B |
Principali anidridi dei gruppi B
Cromo (+2, +3, +6) anidride cromica (nox + 6) CrO3
con nox +2 e +3 forma due ossidi
VII gruppo B |
Manganese (+2, +3, +4, +6, +7) anidride permanganica (nox +7) Mn2O7
con i nox più bassi forma tre ossidi (con nox + 6 i manganati)
Idracidi
Gli idracidi sono composti binari dei non metalli con l'idrogeno.
I principali idracidi si formano dall'unione dell'idrogeno con i non metalli del VII gruppo A (alogeni) e con i non metalli del VI gruppo A.
Il nome degli idracidi si forma facendo seguire al termine "acido" il nome del non metallo seguito dalla desinenza -idrico.
Negli idracidi del VII gruppo A i non metalli presentano sempre nox -1
acido fluoridrico HF
acido cloridrico HCl (acido muriatico)
acido bromidrico HBr
acido Iodidrico HI
Negli idracidi del VI gruppo A i non metalli presentano nox -2
acido solfidrico H2S
acido selenidrico H2Se
acido telluridrico H2Te
Altri idracidi sono
l'acido cianidrico HCN HCN
l'acido azotidrico HN3 HNNN
Altri composti idrogenati binari sono
l'ammoniaca NH3
la fosfina PH3
l'arsina AsH3
Composti ternari: Ossiacidi ed Idrossidi
Ossidi e anidridi reagiscono con l'acqua per dare due importanti classi di composti ternari, gli idrossidi e gli acidi ossigenati o ossiacidi, i quali oltre a contenere ossigeno contengono evidentemente anche idrogeno.
Gli acidi sono sostanze che, sciolte in acqua, tendono a dissociarsi in un anione e in uno o più ioni H+.
Gli idrossidi sono sostanze a carattere basico che, sciolte in acqua, tendono a dissociarsi in un catione e in uno o più anioni ossidrile OH-.
Un composto ternario che contenga idrogeno e ossigeno viene convenzionalmente scritto in modo diverso a seconda che presenti un carattere acido o basico.
Se si tratta di un acido vengono messi in evidenza gli atomi di idrogeno, scrivendo per primo l'idrogeno seguito dal simbolo chimico del non metallo X ed infine dall'ossigeno.
ACIDO HnXmOl
se si tratta di un idrossido vengono messi in evidenza i gruppi ossidrili, scrivendo per primo il simbolo dell'elemento metallico Y seguito da tanti gruppi ossidrili racchiusi tra parentesi tonde, quanti ne richiede il numero di ossidazione "n" del metallo.
IDROSSIDO Y(OH)n
Alcuni composti possono comportarsi come acido o come base, a seconda delle condizioni di reazione. Sono detti composti anfoteri e la loro formula chimica può essere scritta come quella di un acido o come quella di un idrossido in realazione alla particolare comportamento che presentano in una data reazione.
fonte: http://www.pianetachimica.it/didattica/documenti/Chimica_Generale.doc
Chimica generale 6
Visita la nostra pagina principale
Chimica generale 6
Termini d' uso e privacy