
Chimica generale 9
Chimica generale 9
Determinazione dell’ordine di reazione
La determinazione sperimentale degli ordini di reazione segue sostanzialmente tre tecniche. La prima è quella dell’isolamento di uno dei reagenti. La seconda è il metodo delle velocità iniziali. La terza è quella dell’integrazione dell’equazione cinetica.
Metodo dell’isolamento
Questo metodo, introdotto da Ostwald, usa un grande eccesso della concentrazione dei reagenti tranne uno. Per semplicità consideriamo due reagenti A e B per i quali dovrà valere la legge di velocità

Se [B] >> [A] si può ritenere che la concentrazione di B non vari considerevolmente nel tempo e quindi si può concludere che la velocità di reazione sia funzione soltanto della concentrazione di A, scrivendo che

La quantità  è la costante di velocità apparente della reazione. Da questa relazione si determina l’ordine m misurando la velocità di reazione a due concentrazioni di A:
è la costante di velocità apparente della reazione. Da questa relazione si determina l’ordine m misurando la velocità di reazione a due concentrazioni di A:

da cui passando ai logaritmi

Esempio
In corrispondenza del rapporto

si è misurato un rapporto delle velocità pari a

Si ricava quindi l'ordine di reazione

Metodo delle velocità iniziali
Si determina la velocità all’inizio della reazione dalla misura della variazione di concentrazione in un breve intervallo di tempo, viniz = -DA/ Dt. Dato che la concentrazione in questo piccolo intervallo di tempo varia di poco si può considerare approssimativamente uguale a quella iniziale e dunque la legge di velocità si scrive così

Ripetendo l'esperimento con una diversa concentrazione iniziale [A]0' mantenendo invariata la concentrazione iniziale di B si ha

Dal rapporto tra le due misure si ottiene

da cui si determina l'ordine di reazione di A:

Lo stesso procedimento si ripete per determinare l'ordine n di B.
Esempio
Per la reazione in fase gassosa fra ossido di azoto NO e idrogeno H2 si hanno le seguenti misure, della velocità iniziale di reazione con concentrazioni (pressioni parziali) diverse di NO e la medesima concentrazione (pressione parziale) di H2.
NO |
piniz |
v iniz= -Dp/Dt |
|
359 torr |
1.50 torr/s |
|
148 torr |
0.25 torr/s |
L’ordine di reazione m rispetto a NO sarà

Mantenendo invece costante la concentrazione iniziale di NO ed eseguendo la misura della velocità con concentrazioni (pressioni parziali) diverse di H2 si ottengono le seguenti misure
H2 |
piniz |
v iniz= -Dp/Dt |
|
289 torr |
1.60 torr/s |
|
147 torr |
0.79 torr/s |
L’ordine di reazione n rispetto ad H2 sarà

Uso dell’equazione cinetica integrata
Se si possiedono una serie di misure di concentrazione C a tempi diversi è possibili riportarli in un diagramma come ln(C) in funzione del tempo e come 1/C in funzione del tempo. Il grafico che si presenta come una retta permette di discriminare il tipo di cinetica.
Esempio
Il saccarosio, in una soluzione acida diluita si idrolizza per dare glucosio e fruttosio.
I dati sperimentali della velocità della reazione sono raccolti nella seguente tabella:
t (min) |
C = [Saccarosio] mol/L |
Ln C |
1/C |
0 |
0,316 |
-1,152 |
3,16 |
39 |
0,274 |
-1,294 |
3,65 |
80 |
0,238 |
-1,435 |
4,20 |
140 |
0,190 |
-1,661 |
5,26 |
210 |
0,146 |
-1,924 |
6,85 |
Se riportiamo in un grafico il logaritmo della concentrazione in funzione del tempo otteniamo una retta
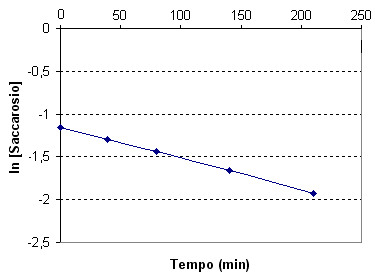
Riportando in un grafico il reciproco della concentrazione in funzione del tempo, si ottiene invece
Costante di velocità specifica ed equazione di Arrhenius
Nel 1889 il chimico svedese Svante Arrhenius propose la seguente relazione per calcolare il valore della costante di velocità specifica

dove
A è una costante che dipende dalla natura dei reagenti
e = 2,718 è la base dei logaritmi naturali o neperiani
Eatt è l'energia di attivazione della reazione
R è la costante universale dei gas
T è la temperatura assoluta
La relazione mostra come la velocità di una reazione, oltre a dipendere naturalmente dal tipo di reazione e dalla natura chimica dei reagenti (ad esempio le reazioni redox sono in genere più lente delle reazioni non-redox), dipende dalla temperatura e da un fattore energetico detto Energia di attivazione.
Il significato fisico della relazione di Arrhenius può essere meglio compreso facendo riferimento alla teoria delle collisioni o teoria degli urti.
Secondo tale teoria affinché le molecole dei reagenti possano produrre una reazione chimica esse devono urtarsi con un'energia cinetica sufficientemente elevata. L'urto deve cioè essere abbastanza violento da rompere i legami che devono essere spezzati affinché avvenga la reazione.
La velocità della reazione dipende quindi dalla frequenza degli urti efficaci (sufficientemente energetici e correttamente orientati). In tal modo possiamo anche spiegarci perché la velocità della reazione aumenta in genere all'aumentare della concentrazione dei reagenti. Infatti una maggior concentrazione aumenta la probabilità che le particelle si urtino e quindi anche la frequenza degli urti tra particelle sufficientemente energetiche.
Ora la meccanica statistica dimostra che, ad una data temperatura, non tutte le particelle possiedono la stessa velocità, ma le velocità si distribuiscono in modo caratteristico secondo una curva di frequenza detta maxwelliana.
L'energia di attivazione può essere quindi definita come la minima energia che deve possedere una particella affinché il suo urto sia efficace e produca una trasformazione chimica.

Dall'osservazione del grafico si può facilmente comprendere come all'aumentare della temperatura aumenta anche la frazione di molecole che possiedono energia superiore all'energia di attivazione. In effetti con l'aumentare della temperatura aumenta in generale la frequenza degli urti tra le molecole, ma in questo caso tale fattore risulta trascurabile rispetto al fattore energetico.
Boltzmann dimostrò che il termine esponenziale  della relazione di Arrhenius esprime effettivamente la frazione di urti che, ad una certa temperatura, presenta energia superiore all'energia di attivazione (urti sufficientemente energetici).
della relazione di Arrhenius esprime effettivamente la frazione di urti che, ad una certa temperatura, presenta energia superiore all'energia di attivazione (urti sufficientemente energetici).
Ad esempio se vogliamo calcolare la fazione di urti sufficientemente energetici a 30°C in una reazione avente Eatt = 15 kJ/mol, otteniamo

in queste condizioni dunque solo 26 urti su 10.000 risultano sufficientemente energetici.
Il fatto che la relazione tra frequenza di urti sufficientemente energetici e temperatura ed energia di attivazione sia di tipo esponenziale ci suggerisce come la velocità di una reazione sia estremamente sensibile alle variazioni di questi due fattori.
Ad esempio se raddoppiamo l'energia di attivazione dell'esempio precedente, portandola a 30 kJ/mol, la frequenza degli urti sufficientemente energetici diventa

circa 66 urti sufficientemente energetici su 10 milioni. La reazione è circa 400 volte più lenta.
In altre parole, a parità di altre condizioni, una reazione che presenta un'energia di attivazione molto elevata risulta essere estremamente lenta. In alcuni casi essa può essere talmente lenta da risultare impercettibile.
Naturalmente un modo per accelerare una tale reazione è quello di aumentare la frazione degli urti sufficientemente energetici, elevando la temperatura.
Se ad esempio aumentiamo la temperatura nella reazione precedente del 50% portandola da 303 K a 454,5 K la frequenza degli urti sufficientemente energetici diventa

188 urti sufficientemente energetici su 10.000, con un aumento della velocità di reazione di oltre il 700%
Si tenga comunque presente che l'aumento della velocità di reazione al crescere della temperatura è tanto più evidente quanto maggiore è l'energia di attivazione. Le reazioni caratterizzate da un'elevata energia di attivazione risultano quindi più sensibili alle variazioni di temperatura.
Nel grafico seguente si può osservare come la curva con Eatt = 100 kJ/mol si impenni in modo più vistoso, al crescere della temperatura di quella con Eatt = 50 kJ/mol.

Il valore dell'energia di attivazione di una reazione compare anche quando rappresentiamo la variazione dell'energia potenziale di una reazione rispetto al tempo. In questo caso possiamo immaginare che l’energia cinetica dei reagenti si trasformi in parte nell’energia potenziale del complesso attivato

Secondo tale modello i reagenti, anche nelle reazioni esoergoniche come quella rappresentata nella figura precedente, devono comunque superare una barriera energetica (che coincide con l'energia di attivazione) per potersi trasformare nei prodotti di reazione. Il modello prevede che durante la reazione si formi un composto instabile, detto complesso attivato o stato di transizione, il cui contenuto energetico risulta più elevato e che condiziona la velocità della reazione stessa.
Nelle reazioni a più stadi deve essere superata più di una barriera energetica (si formano diversi stadi di transizione e dei composti intermedi) e la velocità complessiva della reazione dipende evidentemente dallo stadio più lento.

Il fattore A o fattore pre-esponenziale, risulta essere relativamente costante al variare della temperatura e assume un valore caratteristico per ogni reazione. La teoria degli urti esprime il fattore pre-esponenziale come prodotto di due fattori:
- un fattore di probabilità (o sterico) che dipende essenzialmente dalla geometria delle molecole e misura la probabilità che i reagenti, aventi una certa struttura geometrica, si scontrino con un opportuno orientamento.
- un fattore di frequenza che misura la frequenza degli urti (la probabilità che l’urto avvenga) e dipende dalle dimensioni delle particelle, dalla loro massa e dalla loro temperatura.
Le variazioni di temperatura incidono comunque in modo trascurabile sul fattore pre-esponenziale rispetto a quanto non accada per il fattore energetico o fattore esponenziale (frazione degli urti sufficientemente energetici).
Urti efficaci = urti correttamente orientati x frequenza urti x urti sufficientemente energetici
La relazione di Arrhenius può essere utilizzata per calcolare l'energia di attivazione di molte reazioni. Misurando infatti la costante di velocità specifica di una reazione a due temperature diverse T1 e T2 e dividendo membro a membro, si ottiene

da cui

Ad esempio, sapendo che per la reazione 2N2O → 2N2 + O2, la costante di velocità specifica a 1000 K vale 0,76 s-1, mentre a 1050 K diventa 3,4 s-1, calcoliamo l'energia di attivazione

Altri fattori che influenzano la velocità di una reazione
Finora abbiamo individuato 4 fattori che influiscono sulla velocità di una reazione. Essi sono:
1) la concentrazione
2) la natura chimica dei reagenti ed il tipo di reazione
3) la temperatura
4) l'energia di attivazione (che è caratteristica di ogni reazione)
Esistono infine altri due fattori che influenzano la velocità di una reazione. Essi sono:
6) il tipo di contatto tra reagenti
7) la presenza di catalizzatori
Il tipo di contatto tra reagenti è fondamentale solo nelle reazioni che decorrono in fase eterogenea. Per fase si intende una porzione omogenea di un sistema delimitata da una superficie di separazione fisicamente definita (ad esempio del ghiaccio in acqua forma un sistema bifasico)
Se ad esempio immergiamo del rame metallico in una soluzione di acido nitrico, potremo osservare come la reazione risulta notevolmente accelerata se il rame viene preventivamente polverizzato. Così mentre una barra di ferro può essere riscaldata senza incendiarsi a contatto con l'ossigeno atmosferico, la polvere di ferro nelle stesse condizioni di temperatura brucia vivacemente. La ragione della diversa velocità va ricercata nella maggior superficie di contatto che si produce tra reagenti finemente suddivisi che aumenta enormemente la frequenza degli urti.
Naturalmente nelle reazioni che decorrono in fase omogenea (unica fase), come sono tipicamente quelle in cui tutti i reagenti sono in soluzione o si trovano allo stato gassoso, la superficie di contatto è già la massima possibile in quanto la mescolanza tra i reagenti si produce a livello molecolare. In tal caso evidentemente il tipo di contatto viene sostituito da variazioni nelle concentrazioni che, facendo variare l'affollamento molecolare, influiscono direttamente sulla frequenza degli urti.
I catalizzatori sono sostanze che aggiunte in piccole quantità ad una reazione chimica la accelerano senza venir consumati durante la reazione stessa. Un catalizzatore presenta infatti la caratteristica di trovarsi sempre inalterato alla fine di ogni reazione.
I principi di funzionamento di un catalizzatore sono diversi e non sempre perfettamente chiariti. In generale un catalizzatore permette alle molecole di reagire attraverso un meccanismo differente e più conveniente dal punto di vista energetico. In tal modo, grazie all'aggiunta di un catalizzatore, l'energia di attivazione risulta essere minore di quella originaria e ciò ha come diretta conseguenza che un maggior numero di molecole si trova a possedere un'energia superiore a quella di attivazione.

Equilibrio chimico
La maggior parte delle reazioni chimiche non comporta la completa trasformazione dei reagenti in prodotti, poiché man mano che si formano i prodotti ha luogo la reazione inversa che porta dai prodotti ai reagenti.
Sia ad esempio la reazione
aA + bB  cC + dD
cC + dD
La doppia freccia di reazione indica che si tratta di un equilibrio chimico e che la reazione avviene contemporaneamente nelle due direzioni.
Inizialmente ha luogo solo la reazione diretta, la cui velocità Vdir, essendo proporzionale alla concentrazione dei reagenti, è molto elevata

Mentre la reazione procede le concentrazioni dei reagenti diminuiscono, mentre aumentano le concentrazioni dei prodotti di reazione e si avvia anche la reazione inversa, la cui velocità Vinv sarà naturalmente proporzionale alla concentrazione dei prodotti di reazione

Legge di azione di massa (legge di Guldberg-Waage)
Inizialmente la velocità della reazione diretta sarà maggiore della velocità della reazione inversa (Vdir > Vinv), ma poiché con il procedere della reazione i reagenti diminuiscono ed i prodotti aumentano e le velocità di reazione sono proporzionali alle concentrazioni, la velocità della reazione diretta è destinata a diminuire mentre la velocità della reazione inversa è destinata ad aumentare.

Ad un certo punto si giungerà ad un equilibrio dinamico, cioè al punto in cui la velocità con cui i reagenti si trasformano in prodotti è pari alla velocità con cui i prodotti si trasformano in reagenti. In altre parole

e quindi

dove le concentrazioni delle specie chimiche sono quelle che compaiono al raggiungimento dell'equilibrio. La reazione sembra dunque ferma, anche se in realtà essa continua senza che vi sia una modificazione nelle concentrazioni raggiunte.
Costante di equilibrio (kc kp kn)
La relazione di equilibrio viene data sotto la seguente forma

dove le concentrazioni sono sempre le concentrazioni delle specie chimiche all'equilibrio e kc, essendo ottenuta come rapporto di due costanti (costante di velocità specifica inversa e diretta) è una nuova costante, detta costante di equilibrio. Il suo valore è caratteristico per ogni reazione e varia solo in funzione della temperatura, mentre è indipendente da ogni altra condizione (pressione, concentrazione, catalizzatori etc).
Tale relazione è fondamentale nella descrizione degli equilibri chimici ed è nota come legge di azione di massa o legge di Guldberg-Waage.
La legge di azione di massa afferma dunque che in una reazione che ha raggiunto l'equilibrio, il rapporto tra il prodotto delle concentrazioni dei prodotti e quello dei reagenti, ciascuna elevata al proprio coefficiente stechiometrico è una costante.
Quando la reazione avviene tra sostanze gassose torna utile esprimere la costante di equilibrio come rapporto tra le pressioni parziali. La costante che si ottiene è detta kp.

tenendo conto che la pressione parziale del reagente A vale

e che la stessa relazione vale anche per tutte le altre specie chimiche gassose, possiamo esprimere la kp in funzione delle molarità dei singoli reagenti

Dove Δn è la differenza tra i coefficienti stechiometrici dei prodotti di reazione e i coefficienti stechiometrici dei reagenti. Tale relazione ci permette di affermare che, per una medesima reazione, il valore di kc coincide con il valore di kp solo se reagenti e prodotti sono presenti con lo stesso numero di moli.
È inoltre possibile esprimere la costante di equilibrio in funzione del numero di moli presenti all’equilibrio. La costante che si ottiene è detta kn.

E’ semplice verificare che tra kc e kn esiste la seguente relazione

Il valore assunto dalla costante di equilibrio (kc, kp o kn) ci informa se la reazione avviene in modo più o meno completo.
Se il valore della costante di equilibrio è elevato (in genere molto maggiore di 1) ciò significa che il numeratore è molto più grande del denominatore: l'equilibrio viene cioè raggiunto quando le concentrazioni dei prodotti di reazione sono molto maggiori delle concentrazioni dei reagenti. In tal caso si dice che l'equilibrio è spostato verso destra (verso i prodotti).
Se il valore della costante di equilibrio è basso (in genere molto minore di 1) ciò significa che il numeratore è molto più piccolo del denominatore: l'equilibrio viene cioè raggiunto quando le concentrazioni dei reagenti sono molto maggiori delle concentrazioni dei prodotti di reazione . In tal caso si dice che l'equilibrio è spostato verso sinistra (verso i reagenti).
Si tenga presente che il valore della costante di equilibrio non da alcuna indicazione sulla velocità della reazione, la quale dipende essenzialmente dall'energia di attivazione, dalla temperatura e dalle concentrazioni dei reagenti.
I valori delle costanti di equilibrio si trovano tabulati per le diverse reazioni. Conoscendole possiamo ricavare le concentrazioni delle specie chimiche all'equilibrio. L’unità di misura delle costanti di equilibrio varia a seconda della stechiometria della reazione ed in genere sarà pari per la kc a (mol l-1)Dn e per la kp a (atm l-1)Dn
Esempio 1
Introduciamo 2 moli di H2 e 0,8 moli di I2 in un recipiente di 1,6 litri e portiamo la temperatura a 763 K per produrre le reazione
H2 + I2  2HI
2HI
sapendo che a 763 K la kc = 46 si calcolino le concentrazioni di equilibrio.
Se indichiamo con x le moli di idrogeno che reagiscono con x moli di iodio per litro, all'equilibrio si formeranno 2x mol/L di acido iodidrico. Costruiamo allora la seguente tabella
Specie |
Concentrazioni iniziali |
Concentrazioni |
H2 |
2/1,6 = 1,25 mol/L |
1,25 - x mol/L |
I2 |
0,8/1,6 = 0,5 mol/L |
0,5 - x mol/L |
HI |
0 mol/L |
2x mol/L |
Utilizziamo ora i valori delle concentrazioni di equilibrio, espresse in funzione di x, all'interno delle legge di azione di massa

e, sostituendo opportunamente le concentrazioni di equilibrio

si ottiene un'equazione di secondo grado che risolta fornisce le seguenti due radici
x1 = 1,442 x2 = 0,475
La prima va scartata non avendo significato fisico (è superiore alla concentrazione iniziale).
Le concentrazioni di equilibrio saranno pertanto pari a
Specie |
Concentrazioni |
H2 |
1,25 – x = 1,25 – 0,475 = 0.775 mol/L |
I2 |
0,5 – x = 0,5 – 0,475 = 0.025 mol/L |
HI |
2x = 2(0,475) = 0,95 mol/L |
Esempio 2
In un recipiente dal volume di 11 litri si introducono a temperatura ambiente 0,2 moli di A2B e 0,04 moli di B2. Il sistema viene portato alla temperatura di 750°C e si instaura il seguente equilibrio gassoso:
2A2B(g) D 2A2(g) + B2(g)
La pressione all'equilibrio risulta pari a 2,29 atm. Calcoliamo la kp e la Kc a 750°C.
Poiché durante la reazione, per ogni 2 moli di A2B che reagiscono se ne formano 2 di A2 ed 1 di B2, potremmo affermare che, all’equilibrio, il numero di moli di A2B è diminuita di 2x, quello di A2 è aumentato di 2x, mentre quello di B2 è aumentato di x unità.
Il numero di moli delle tre specie chimiche all’equilibrio sarà pertanto pari a
nA2B = 0,2 – 2x
nA2 = 2x
nB2 = 0,04 + x
All’equilibrio saranno pertanto presenti nel contenitore un numero di moli totali pari a
ntot = nA2B + nA2 + nB2 = (0,2 – 2x) + 2x + (0,04 + x) = 0,24 + x
Queste (0,24 + x) moli generano, a T = 750 + 273,15 = 1023,15 K. una pressione complessiva pari 2,29 atmosfere. Scriviamo allora l’equazione di stato per la miscela gassosa all’equilibrio


da quest’ultima relazione possiamo ricavare la x che vale
x = 0,06 mol
Il numero di moli all’equilibrio delle tre specie chimiche sarà pertanto
nA2B = 0,2 – 2x = 0,2 -2(0,06) = 0,08
nA2 = 2x = 2(0,06) = 0,12
nB2 = 0,04 + x = 0,04 + 0,06 = 0,10
Il numero totale di moli della miscela gassosa all’equilibrio è dunque pari a 0,08 + 0,12 + 0,10 = 0,30.
Calcoliamo la kn dell’equilibrio

Calcoliamo ora la Kc usando la relazione che lega le due costanti Kc = Kn / VΔn, dove Δn è la differenza tra i coefficienti dei prodotti (2+1=3) e dei reagenti (2). Poiché in questo caso Δn = 3 - 2 = 1, avremo

Calcoliamo ora la Kp usando la relazione che lega le due costanti Kc = Kp / (RT)Δn

Esempio 3
Un reattore di 6 litri, in cui è stato fatto il vuoto, viene caricato con 80 mmol del composto AB e portato a 350° C. A questa temperatura si instaura il seguente equilibrio:
AB(g) D A(g) + B(g)
Sapendo che la Kp a 350°C è 2,70 atm, calcoliamo la pressione parziale di ogni singolo componente all'equilibrio, la pressione totale della miscela all’equilibrio e la sua composizione percentuale in volume.
A) Calcoliamo la pressione iniziale del composto AB applicando l’equazione di stato dei gas

B) Calcoliamo le pressioni parziali e la pressione totale della miscela all’equilibrio
All’equilibrio x moli di AB avranno reagito per dare x moli di A e x moli di B. Poiché la pressione è proporzionale al numero di moli, potremo affermare che anche la pressione parziale dei singoli componenti è variata nelle medesime proporzioni in cui è variato il loro numero di moli.
Pertanto all’equilibrio le pressioni parziali dei tre componenti saranno
PAB = 0,68 – x
PA = x
PB = x
Scriviamo la relazione di equilibrio (Guldberg-Waage) rispetto alle pressioni parziali.

Riordinando si ottiene la seguente equazione di secondo grado
x2+2,7x-1,836=0
che, risolta, fornisce le seguenti radici
x1 = 0,563 x2 = -3,263
Scartiamo la seconda radice poiché non ha significato fisico (la concentrazione del reagente all’equilibrio dovrebbe essere più elevata rispetto a quella iniziale). Le pressioni parziali all’equilibrio saranno pertanto
PAB = 0,68 – x = 0,68 – 0,563 = 0,117 atm
PA = + x = 0,563 atm
PB = + x = 0,563 atm
All’equilibrio il sistema avrà dunque una pressione totale pari alla somma delle pressioni parziali dei singoli componenti (Legge di Dalton delle pressioni parziali)
Ptot = PAB + PA + PB = 0.117 + 0,563 + 0,563 = 1,243 atm
C) Calcoliamo la composizione percentuale in volume della miscela all’equilibrio
La composizione in volume è la medesima della frazione molare, poiché i volumi sono proporzionali al numero di moli.
Per calcolare la frazione molare dei singoli componenti all’equilibrio è sufficiente ricordare che, per la legge di Dalton delle miscele gassose, La frazione molare è pari al rapporto tra pressione parziale e pressione totale (il numero di moli di un componente sta infatti al numero di moli totali della miscela come la pressione parziale del componente sta alla pressione totale)



Dunque se potessimo separare i 3 gas all’interno del recipiente, il composto AB che è presente con 94 molecole ogni 1000 molecole di miscela occuperebbe il 9,4% del volume, mentre il composto A ed il composto B, che sono presenti con 453 molecole ciascuno ogni 1000 molecole di miscela, occuperebbero ognuno il 45,3% del volume.
Equilibri chimici omogenei ed eterogenei
Si parla di equilibri in fase omogenea quando tutte le specie chimiche coinvolte nella reazione sono presenti in un'unica fase. Si definisce fase una porzione omogenea di un sistema, delimitata da una superficie di separazione fisicamente definita. Così ad esempio del ghiaccio in acqua liquida costituisce un sistema bifasico, mentre una soluzione è un sistema in fase unica. Sono tipicamente omogenei gli equilibri che decorrono in fase gassosa e quelli in soluzione.
Si parla invece di equilibri in fase eterogenea quando almeno una delle specie chimiche coinvolte nella reazione si trova in una fase diversa dalle altre. In tal caso risulta conveniente far comparire nella relazione di equilibrio solo le concentrazioni delle specie chimiche le cui concentrazioni possono variare in funzione delle condizioni sperimentali (in pratica le specie chimiche allo stato gassoso e i soluti).
Si tenga infatti presente che la concentrazione di un solido o un liquido allo stato puro è una costante.
Proviamo ad esempio a calcolare la molarità di un campione di ferro del peso di 50 g sapendo che la densità del ferro è pari a 7860 g/dm3 ed il suo peso molare è pari a 55,85 g/mol.

Come si può osservare la molarità è indipendente dal peso (W) del campione considerato. Infatti al crescere del peso del campione crescono proporzionalmente sia il numero di moli che il volume, in modo che il loro rapporto rimane comunque costante.
Tenendo conto di quanto detto, si è convenuto che, qualora in un equilibrio eterogeneo la concentrazione di una specie chimica risulti costante, essa vada inglobata nella costante di equilibrio.
Se ad esempio facciamo reagire della polvere di grafite solida, con dell'ossigeno gassoso per ottenere dell'ossido di carbonio, secondo la reazione
2C(s) + O2(g)  2CO(g)
2CO(g)
La relazione di equilibrio risulta essere

poiché però la concentrazione del carbonio solido è una costante si avrà

Esempio
A 1200 K il carbonato di calcio si decompone in ossido di calcio e anidride carbonica con una kp = 4,5. Dopo aver introdotto 80 g di carbonato in un recipiente di 10 L a 1200 K , calcolare la pressione prodotta dall'anidride carbonica e la massa indecomposta del carbonato all'equilibrio.
CaCO3(s) D CaO(s) + CO2(g)
Tenendo conto che sia il carbonato che l'ossido di calcio sono solidi, la relazione di equilibrio sarà

La pressione esercitata dall'anidride carbonica all'equilibrio è dunque di 4,5 atm
Calcoliamo ora quante moli di anidride carbonica devono essere presenti in un recipiente di 10 litri a 1200 K per produrre una pressione di 4,5 atmosfere.

poiché ciascuna mole di carbonato che reagisce produce 1 mole di ossido ed 1 di anidride, possiamo dedurre che, se si sono formate 0,46 moli di CO2, si devono essere decomposte altrettante moli di carbonato.
Poiché il peso molare del carbonato di calcio è di 100 g/mol, siamo in grado di calcolare quanti grammi di carbonato hanno reagito

Rimarranno dunque indecomposti, una volta raggiunto l'equilibrio, (80 - 46) = 34 grammi di carbonato di calcio.
Modificazioni di un equilibrio chimico: il principio di Le Chatelier
Di particolare interesse pratico nello studio degli equilibri chimici è l'analisi dei fattori che in qualche modo possano influire sull'equilibrio, spostandolo verso le specie chimiche che si desidera ottenere.
Il principio di Le Chatelier, o principio dell’equilibrio mobile, ci offre un criterio generale per prevedere lo spostamento di un equilibrio in risposta a sollecitazioni esterne.
Il principio afferma infatti che un sistema in equilibrio tende a mantenerlo inalterato, neutralizzando per quanto possibile qualsiasi azione di disturbo esterna.
Per quanto riguarda un equilibrio chimico possiamo affermare che se esso viene sottoposto ad un'azione perturbatrice esterna, l'equilibrio si sposterà, facendo variare le concentrazioni di equilibrio delle specie chimiche, in modo tale da rendere minimi gli effetti della perturbazione.
Prima di analizzare le diverse perturbazioni cui può essere sottoposto un equilibrio chimico e gli spostamenti relativi, prevedibili sulla base del principio di Le Chatelier, ricordiamo che il valore della costante di equilibrio viene modificato solo da variazioni della temperatura, mentre rimane costante per ogni altra modificazione delle condizioni sperimentali.
1) Modificazione delle concentrazioni
Applicando il principio di Le Chatelier possiamo prevedere che in risposta ad una variazione nella concentrazione di una delle specie chimiche che partecipa alla reazione, l'equilibrio si sposti in modo da riottenere la concentrazione originaria.
In altre parole se aumentiamo la concentrazione di una specie chimica l'equilibrio si sposterà dalla parte opposta, se invece diminuiamo la concentrazione di una specie chimica l'equilibrio si sposterà verso il lato della reazione in cui è presente la specie la cui concentrazione è diminuita.
Per esemplificare quanto detto ricalcoliamo le concentrazioni di equilibrio per la reazione
H2 + I2  2HI
2HI
nell'ipotesi che all’equilibrio vengano aggiunte 0,4 moli di Iodio. Nell’esempio precedente erano inizialmente presenti 0,8 moli di Iodio. Sarà allora sufficiente ricalcolare le concentrazioni di equilibrio come se inizialmente fossero presenti 0,8 + 0,4 = 1,2 moli di Iodio. L’equilibrio finale non dipende infatti dal momento in cui lo disturbiamo (possiamo pensare di aggiungere 0,4 moli indifferentemente all’inizio o alla fine della reazione). Costruiamo allora una nuova tabella
Specie |
Concentrazioni iniziali |
Concentrazioni |
H2 |
2/1,6 = 1,25 mol/L |
1,25 - x mol/L |
I2 |
1,2/1,6 = 0,75 mol/L |
0,75 - x mol/L |
HI |
0 mol/L |
2x mol/L |
Utilizziamo ora i valori delle concentrazioni di equilibrio, espresse in funzione di x, all'interno delle legge di azione di massa

e, sostituendo opportunamente le concentrazioni di equilibrio

si ottiene un'equazione di secondo grado che risolta fornisce la seguente soluzione
x = 0,68
Le nuove concentrazioni di equilibrio saranno pertanto pari a
Specie |
Concentrazioni |
H2 |
1,25 – x = 1,25 – 0,68 = 0.57 mol/L |
I2 |
0,75 – x = 0,75 – 0,68 = 0.07 mol/L |
HI |
2x = 2(0,68) = 1,36 mol/L |
Se le confrontiamo con le concentrazioni di equilibrio precedenti l’aggiunta di 0.4 moli di Iodio, osserviamo come l’ l'equilibrio si sia spostato verso destra, cercando in questo modo di diminuire la concentrazione del reagente I2, che era stata aumentata. Parte dello iodio aggiunto ha reagito con l’idrogeno (facendone diminuire la concentrazione) per dare acido iodidrico (aumentandone la concentrazione).
Specie |
Concentrazioni |
Concentrazioni |
Variazione |
H2 |
0.775 mol/L |
0.57 mol/L |
– |
I2 |
0.025 mol/L |
0.07 mol/L |
+ |
HI |
0,95 mol/L |
1,36 mol/L |
+ |
Un modo per far avvenire completamente una reazione è ad esempio quello di eliminare continuamente i prodotti di reazione mentre si formano (non sempre è comunque possibile). In questo modo infatti la reazione si sposta continuamente verso destra fino a che tutti i reagenti non si sono trasformati in prodotti di reazione, senza essere mai in grado di raggiungere l'equilibrio.
Un altro modo di spostare una reazione verso i prodotti di reazione è di farla avvenire con un eccesso di un reagente sugli altri.
Esempio
In un reattore nel quale vengono introdotti 4 g di PCl5 si instaura il seguente equilibrio omogeneo in fase gassosa:
PCl5 D PCl3 + Cl2
con formazione di 0.8 g di PCl3. Calcoliamo i grammi di Cl2 da inserire nel reattore per ridurre la quantità di PCl3 a 0.5 g.
A) Calcoliamo il numero di moli delle tre specie chimiche all’equilibrio.
I pesi molari sono
PCl5 = 208.25 g/mol PCl3= 137.35 g/mol Cl2 = 70.90 g/mol
Il numero di moli iniziali di PCl5 è pari a 4/208.25 = 1.921x10-2
Il numero di moli di PCl3 che si formano all’equilibrio è pari a 0.8/137.35 = 5.82x10-3.
Poiché durante la reazione per ogni x moli di PCl5 che reagiscono si formano altrettante moli di PCl3 e di Cl2, allora 5.82x10-3 sono anche le moli di PCl5 che hanno reagito e le moli di Cl2 che si sono formate all’equilibrio. Il numero di moli delle tre specie chimiche all’equilibrio sarà pertanto
PCl5 = 1.921x10-2 - 5.82x10-3 = 1.339x 10-2
PCl3 = 5.82x10-3
Cl2 = 5.82x10-3
B) Calcoliamo la Kn della reazione

Immaginiamo ora di aggiungere all’equilibrio x moli di Cl2. In queste condizioni l’equilibrio si sposta verso sinistra (principio di Le Chatelier). Delle x moli di Cl2 che abbiamo aggiunto, una parte y reagirà con y moli di PCl3 per dare y moli di PCl5 . In questa situazione il numero di moli all’equilibrio saranno
PCl5 D PCl3 + Cl2
1.339x10-2 + y 5.82x10-3 - y 5.82x10-3 + x – y
La quantità y deve essere tale per cui il numero di moli di PCl3 che rimarranno alla fine siano pari alla quantità desiderata, cioè 0.5/137.35 = 3.64x10-3 mol. Dunque deve essere
5.82x10-3 – y = 3.64x10-3 da cui y = 2.18x10-3
Sostituendo il valore della y trovato si avrà
PCl5 D PCl3 + Cl2
1.339x10-2 + y 5.82x10-3 - y 5.82x10-3 + x - y
1.339x10-2 + 2.18x10-3 5.82x10-3 - 2.18x10-3 5.82x10-3 + x - 2.18x10-3
1.557x10-2 3.64x10-3 3.64x10-3 + x
Sostituendo queste quantità nella relazione di equilibrio si ha

da cui, risolvendo rispetto alla x, si ottiene x = 7.18 10-3 mol, pari a 7.18 10-3 x 70.90 = 0.51 g di Cl2
2) Modificazione della pressione
Le modificazioni della pressione incidono solo sulle reazioni che decorrono in fase gassosa, in quanto liquidi e solidi sono praticamente incomprimibili.
In base al principio di Le Chatelier possiamo prevedere che una reazione in fase gassosa reagisca ad un aumento di pressione esterna spostando il suo equilibrio in modo da rendere minimo tale aumento. In altre parole l'equilibrio si sposterà in modo da ridurre il numero complessivo di molecole presenti all'equilibrio (la pressione è infatti direttamente proporzionale al numero di particelle presenti) e quindi verso il lato della reazione in cui è complessivamente minore il numero di moli gassose.
Da quanto detto risulta evidente che risentono di variazioni di pressione solo le reazioni gassose in cui il numero totale di moli dei reagenti è diverso dal numero totale di moli dei prodotti di reazione. Nel caso in cui il numero di moli gassose dei reagenti sia uguale al numero di moli gassose dei prodotti l’equilibrio risulta indifferente ad un cambiamento di pressione
Prendiamo ad esempio i seguenti tre equilibri gassosi e sottoponiamoli idealmente ad un aumento di pressione
aumento di pressione
N2 + 3H2  2NH3 spostamento verso destra
2NH3 spostamento verso destra
PCl5  PCl3 + Cl2 spostamento verso sinistra
PCl3 + Cl2 spostamento verso sinistra
2HI  H2 + I2 indifferente
H2 + I2 indifferente
Esempio
In un recipiente di 10 litri vengono introdotte 0,8 moli di N2O4 (ipoazotide). Alla temperatura di 299 K si stabilisce il seguente equilibrio
N2O4(g) D 2NO2(g)
la cui costante alla suddetta temperatura è kp = 0,172. Calcolare come varia la concentrazione di equilibrio del biossido di azoto dopo aver portato il volume del recipiente da 10 litri a 2 litri, mantenendo costante la temperatura.
Calcoliamo le pressioni iniziale dell'ipoazotide

Calcoliamo ora la pressione di equilibrio del biossido di azoto, osservando che per x moli di N2O4 che reagiscono si formano 2x moli di biossido e ricordando che le variazioni di pressione sono direttamente proporzionali alle variazioni nel numero di moli.
Specie |
Pressioni iniziali |
Pressioni |
N2O4 |
1,96 atm |
1,96 - x atm |
NO2 |
0 atm |
2 x atm |
Utilizziamo ora i valori delle pressioni di equilibrio, espresse in funzione di x, all'interno delle legge di azione di massa

e sostituendo opportunamente

che, risolta, da il seguente valore x = 0,27 atm
La pressione parziale delle specie chimiche all'equilibrio è dunque pari a
Specie |
Pressioni |
N2O4 |
1,96 – x = 1,96 – 0,27 = 1,69 atm |
NO2 |
2 x = 2(0,27) = 0,54 atm |
Osserviamo come la pressione dell'ipoazotide sia all'equilibrio circa 3 volte maggiore di quella del biossido di azoto.
Calcoliamo ora le pressioni parziali di equilibrio dopo che il volume è stato portato da 10 a 2 litri, con un relativo aumento della pressione esercitata sulla miscela gassosa.
La nuova pressione iniziale per l'ipoazotide sarà ora pari a

L'equazione di equilibrio diventa

x vale ora 0,63 e le nuove pressioni di equilibrio saranno
Specie |
Pressioni |
N2O4 |
9,81 – x = 9,81 – 0,63 = 9,18 atm |
NO2 |
2 x = 2(0,63) = 1,26 atm |
Osserviamo come, dopo aver compresso la miscela gassosa, la pressione dell'ipoazotide sia ora circa 7 volte maggiore di quella del biossido. L'equilibrio si è dunque spostato verso sinistra, dove minore era il numero di moli.
3) Variazione della temperatura
In base al principio di Le Chatelier una reazione reagisce ad un aumento di temperatura modificando le condizioni di equilibrio al fine di rendere minimo l'effetto dell'apporto di calore.
Lo spostamento sarà quindi differente a seconda che la reazione sia esotermica o endotermica.
Per prevedere in modo semplice le variazioni dell'equilibrio è possibile trattare il calore di reazione come un reagente nelle reazioni endotermiche e come un prodotto di reazione nelle reazioni esotermiche.
reazione endotermica A + B + calore  C + D
C + D
reazione esotermica A1 + B1  C1 + D1 + calore
C1 + D1 + calore
- In tal modo se aumentiamo la temperatura, fornendo calore ad una reazione endotermica, la reazione si sposterà verso i prodotti di reazione, poiché in tal modo il calore fornito viene assorbito per formare i composti più energetici. Se aumentiamo invece la temperatura in una reazione esotermica, l'equilibrio si sposta, per lo stesso motivo, verso i reagenti.
- Una diminuzione di temperatura sposta invece i due equilibri in senso opposto, verso il lato in cui compare il calore. In tal modo la reazione si oppone alla diminuzione di temperatura producendo calore.
Le variazioni di temperatura modificano anche il valore della costante di equilibrio, la quale assumerà pertanto valori più elevati se l'equilibrio si sposta verso destra e valori minori se l'equilibrio si sposta verso sinistra.
Tale comportamento può essere interpretato ricordando che la costante di equilibrio si ottiene come rapporto tra le costanti di velocità della reazione diretta e della reazione inversa.
Nel grafico che segue si può osservare come in una reazione esotermica l'energia di attivazione della reazione diretta è maggiore dell'energia di attivazione della reazione inversa.
Come abbiamo già avuto modo di dire, quanto più elevata è l'energia di attivazione tanto più sensibile risulta la velocità di una reazione agli aumenti di temperatura. Per questo motivo un aumento di temperatura accelera maggiormente la reazione inversa (con grande Eatt) della reazione diretta (con piccola Eatt). Ciò implica che kinv aumenta di più di kdir ed il loro rapporto (kc) a temperature maggiori risulta pertanto più piccolo.

Molto spesso è necessario scegliere con grande attenzione le condizioni in cui far avvenire una reazione, poiché facendo variare certi parametri possono ottenersi benefici in termini di resa di una reazione, pagandoli però in termini di velocità. Un tipico esempio di quanto affermato è il processo Haber-Bosch per la produzione dell'ammoniaca, a partire da idrogeno e azoto gassosi
NH2(g) + 3H2(g)  2NH3(g) + 46 kJ/mol
2NH3(g) + 46 kJ/mol
Come si può osservare il processo è esotermico e se vogliamo aumentare la resa di ammoniaca dobbiamo lavorare a basse temperature per spostare l'equilibrio verso destra. In tal modo però la velocità di reazione diventa talmente bassa da risultare economicamente inaccettabile. Aumentando la temperatura aumenta la velocità di reazione, ma l'equilibrio si sposta verso sinistra e la resa in ammoniaca diminuisce drasticamente. La soluzione, proposta da Haber, consiste nel mantenere elevata la temperatura per consentire una velocità di reazione accettabile e di spostare l'equilibrio verso destra, per aumentare la resa, lavorando a pressioni elevate ( la reazione si svolge infatti in fase gassosa ed i prodotti di reazione sono presenti con un numero di moli inferiore rispetto ai reagenti).
Equilibri di dissociazione ionica
Il prodotto ionico dell’acqua
L'acqua pura presenta una piccolissima percentuale di molecole dissociate in ioni H+ e ioni OH- secondo il seguente equilibrio (autoprotolisi o autoionizzazione)
H2O + H2O  OH¯ + H3O+
OH¯ + H3O+
L’equilibrio viene tuttavia descritto in modo semplificato considerando solo la molecola d’acqua che si dissocia e non quella che forma il legame dativo con lo ione H+ per dare lo ione ossonio H3O+
H2O  H+ + OH-
H+ + OH-
Anche per tale reazione di dissociazione è possibile calcolare una costante di equilibrio che, alla temperatura di 25°C, vale

Dal valore della costante di dissociazione deduciamo che l'equilibrio è fortemente spostato verso sinistra. Per questo motivo, possiamo ritenere trascurabile la frazione x di molecole d'acqua che si dissociano rispetto all'acqua indissociata, e considerare la concentrazione di quest'ultima pari alla concentrazione dell'acqua pura.

La concentrazione dell'acqua pura è ovviamente una costante e vale

Si conviene pertanto di inglobare la concentrazione dell'acqua nella costante di dissociazione, ottenendo

La nuova costante kw è detta prodotto ionico dell'acqua e vale

Poiché nell'acqua pura le uniche molecole che si dissociano sono ovviamente quelle dell'acqua e ogni molecola d'acqua che si dissocia produce uno ione H+ ed uno ione OH-, è evidente che le due specie ioniche dovranno trovarsi nell'acqua in numero uguale, dovranno cioè possedere la stessa concentrazione. Risulta pertanto evidente che la loro concentrazione dovrà essere pari a

10-7 è evidentemente anche il numero di moli di acqua che si dissociano in un litro d'acqua. Possiamo pertanto calcolare il suo grado di dissociazione

il che significa che nell'acqua pura a 25°C si dissociano circa 2 molecole d'acqua su 1 miliardo.
Le soluzioni in cui  sono dette neutre
sono dette neutre
Le soluzioni in cui  sono dette acide
sono dette acide
Le soluzioni in cui  sono dette basiche
sono dette basiche
Si tenga comunque presente che la reazione di dissociazione dell'acqua è una reazione endotermica e quindi, in base al principio di Le Chatelier, la kw aumenta all'aumentare della temperatura. Così a temperature maggiori di 25°C la neutralità si raggiunge per concentrazioni degli ioni H+ e OH- leggermente superiori di 10-7 mol/L ( ).
).
Poiché a Temperatura costante kw è costante, si osservi come nel caso sia nota la [H+] rimanga univocamente determinata anche [OH-] e viceversa.
pH e pOH
Essendo [H+] e [OH-] espresse da valori molto piccoli risulta più comodo usare, per misurarle, una notazione logaritmica. Si conviene pertanto di esprimere la concentrazione degli ioni H+ in termini di pH, il quale risulta definito tramite la seguente relazione:

In modo del tutto analogo si può definire come unità di misura della concentrazione degli ioni OH- in una soluzione il pOH

Tra pH e pOH esiste una semplice relazione che possiamo ottenere calcolando il logaritmo negativo di entrambi i membri del prodotto ionico dell'acqua

da cui

La somma del pH e del pOH è sempre uguale a 14
Poiché nelle soluzioni neutre  , allora per esse vale anche pH = pOH = 7
, allora per esse vale anche pH = pOH = 7
Costruiamo ora una tabella che metta in relazione il valore delle concentrazioni degli ioni H+ e degli ioni OH- con i valori del pH e del pOH
[H+] |
pH |
[OH-] |
pOH |
10-15 |
15 |
101 |
-1 |
10-14 |
14 |
100 |
0 |
10-13 |
13 |
10-1 |
1 |
10-12 |
12 |
10-2 |
2 |
10-11 |
11 |
10-3 |
3 |
10-10 |
10 |
10-4 |
4 |
10-9 |
9 |
10-5 |
5 |
10-8 |
8 |
10-6 |
6 |
10-7 |
7 |
10-7 |
7 |
10-6 |
6 |
10-8 |
8 |
10-5 |
5 |
10-9 |
9 |
10-4 |
4 |
10-10 |
10 |
10-3 |
3 |
10-11 |
11 |
10-2 |
2 |
10-12 |
12 |
10-1 |
1 |
10-13 |
13 |
100 |
0 |
10-14 |
14 |
101 |
-1 |
10-15 |
15 |
Come si può notare il pH può assumere anche valori negativi e valori superiori a 14. Si tratta comunque di casi piuttosto rari con concentrazioni di ioni H+ eccezionalmente basse o elevate.
Si noti inoltre come, essendo la scala del pH una scala logaritmica, ogni grado di pH corrisponde ad una variazione nella concentrazione degli ioni H+ pari a 10 volte. Così una soluzione a pH 2 presenta una concentrazione degli ioni H+ 1000 volte maggiore di una soluzione a pH 5.
Calcolo del pH
Calcolo pH per acidi e basi forti
Il calcolo del pH per soluzioni contenenti acidi e basi forti non presenta difficoltà, se naturalmente si conosce la concentrazione iniziale della soluzione. Infatti, poiché gli acidi e le basi forti in acqua sono completamente dissociati, la concentrazione degli ioni H+ (per gli acidi) e degli ioni OH- (per le basi) risultano uguali alla concentrazione iniziale.
Esempi
- Calcolare il pH di una soluzione 10-2 M di HCl.
Poiché l'acido cloridrico è un acido forte esso è completamente dissociato in 10-2 mol/L di ioni H+ e 10-2 mol/L di ioni Cl-. Il pH sarà pertanto pari a

- Calcolare il pH di una soluzione 3.10-5 M di NaOH
Poiché l'idrossido di sodio è una base forte esso è completamente dissociato in 3.10-5 mol/L di ioni OH- e 3.10-5 mol/L di ioni Na+. Il pH sarà pertanto pari a

- Calcolare il pH di una soluzione 5.10-4 M di Ba(OH)2
Poiché l'idrossido di bario si dissocia completamente in uno ione Ba2+ e 2 ioni OH-, in tal caso la concentrazione finale degli ioni OH- sarà doppia della concentrazione iniziale dell'idrossido e pari a 10-3 mol/L. La concentrazione degli ioni H+ sarà quindi pari a 10-11 ed il pH uguale a 11.
pH in soluzioni molto diluite di acidi (e basi) forti
Proviamo a calcolare il pH di una soluzione 10-7 M di HCl. Applicando quanto detto in precedenza il pH dovrebbe essere pari a 7. Si arriva cioè al risultato assurdo e paradossale che una soluzione che contiene un acido forte (per quanto molto diluito) è neutra.
In effetti quando la concentrazione di un acido o di una base forte scende sotto le 10-6 mol/L non è più possibile trascurare gli ioni H+ provenienti dalla dissociazione dell'acqua, che, per l'acqua pura sappiamo essere 10-7 mol/L .
E' quindi necessario in questo caso prendere in considerazione contemporaneamente i due equilibri e sommare gli ioni H+ provenienti dall'acido e quelli provenienti dall'acqua
HCl → H+ + Cl-
H2O  H+ + OH-
H+ + OH-
Naturalmente non è possibile semplicemente sommare i 10-7 ioni H+ provenienti dall'acido con i 10-7 ioni H+ provenienti dall'acqua pura, infatti mentre l'acido forte rimane completamente dissociato, l'acqua, in presenza dei 10-7 ioni H+ provenienti dall'acido, sposta il suo equilibrio verso sinistra, in risposta all'aumentata concentrazione di uno dei suoi prodotti di reazione (H+). L'apporto di ioni H+ dell'acqua sarà dunque minore di 10-7 mol/L.
Se indichiamo con x gli ioni OH- provenienti dalla dissociazione dell'acqua, gli ioni H+ complessivamente in soluzione saranno dati da x ioni provenienti dall'acqua più 10-7 ioni provenienti dall'acido. Poiché tali concentrazioni devono soddisfare al prodotto ionico dell'acqua potremo scrivere

risolvendo l'equazione di 2° grado si ottiene
x = [OH-] = 6,18*10-8 mol/L [H+] = x+ 10-7 = 1,62*10-7
ed il pH risulta perciò pari a

Lo stesso risultato poteva essere ottenuto impostando un sistema di due equazioni con incognite [OH–] e [H+] .

dove la prima equazione è la condizione di equilibrio per la reazione di dissociazione dell'acqua (prodotto ionico) e la seconda è la cosiddetta condizione di elettroneutralità, per cui la soluzione deve essere complessivamente neutra e la somma delle cariche positive deve sempre essere pari alla somma delle cariche negative (bilancio di carica). Si osservi che [Cl-] non è un'incognita, ma vale in questo caso 10-7 mol/L derivando dalla completa dissociazione dell'acido. Dunque, per un acido forte di concentrazione CA, il sistema diventa

esplicitando [OH–] dalla prima equazione e sostituendo nella seconda si ottiene

e, riordinando

un’equazione di secondo grado che ci fornisce, nota la concentrazione iniziale CA, la concentrazione di ioni H+ di una soluzione qualsiasi di un’acido forte. Risolvendo rispetto alla [H+] si ottiene la seguente formula.

mentre per una base forte diluita di concentrazione CB il sistema di equazioni è

ed esplicitando [OH–] dalla prima equazione e sostituendo nella seconda


un’equazione di secondo grado la cui formula risolutiva è

pH in soluzioni di acidi e basi deboli: ka e kb (pka e pkb)
Per il calcolo del pH di soluzioni di acidi e basi deboli non è sufficiente conoscere la loro concentrazione iniziale, in quanto gli acidi deboli non sono completamente dissociati in soluzione acquosa. Per determinare che concentrazione assumeranno gli ioni H+ (o OH–) è quindi necessario conoscere anche il valore della costante dell'equilibrio di dissociazione o costante di dissociazione.
Per un generico acido monoprotico HA, l'equilibrio di dissociazione è
HA  H+ + A–
H+ + A–
La costante di equilibrio, nota come costante di dissociazione acida (o kappa acida) ka, vale

In analogia con la definizione di pH, anche per la costante di dissociazione acida si utilizza il pka.
pka = – log10 (ka)
Per gli acidi deboli poliprotici vi sono naturalmente tante costanti di dissociazione quanti sono gli atomi di idrogeno dissociabili (costante di prima dissociazione  , etc)
, etc)
Ad esempio per l'acido solforoso a 25°C si ha
H2SO3 
HSO3- 
Naturalmente l'acido cede più facilmente il primo ione H+, mentre il secondo ione H+, che deve abbandonare uno ione negativo ed è quindi trattenuto con maggior forza, si separa con maggior difficoltà.
A conferma di quanto detto si può notare come nell'esempio riportato il valore di  . Il primo equilibrio di dissociazione è quindi più spostato verso destra del secondo.
. Il primo equilibrio di dissociazione è quindi più spostato verso destra del secondo.
Si tratta di un comportamento generale. Tutti gli acidi deboli poliprotici presentano infatti valori decrescenti per le costanti di dissociazione successive alla prima.
Quanto detto per gli acidi deboli vale anche per le basi deboli. Ad esempio per una generica base BOH, l'equilibrio di dissociazione è
BOH  B+ + OH –
B+ + OH –
La costante di equilibrio, nota come costante di dissociazione basica (o kappa basica) kb, vale

Naturalmente anche per le basi deboli vi possono essere tante kb quanti sono i gruppi ossidrili dissociabili. Ovviamente, anche la costante di dissociazione basica può essere espressa come pkb.
pkb = – log10 (kb)
Il valore assunto dalla costante di dissociazione è utilizzato come una misura della forza di un acido (o di una base), in quanto è indipendente dalla concentrazione iniziale dell'elettrolita. Possiamo in altre parole affermare che un acido (o una base) è tanto più debole quanto più basso è il valore della sua costante di dissociazione.
Ad esempio l'acido ipocloroso, HClO (ka = 2,95.10-8) è più debole dell'acido fluoridrico HF (ka = 3,53.10-4).
Il grado di dissociazione a di un acido (o di una base) non può essere usato come misura della sua forza in quanto si può facilmente dimostrare che esso varia con la concentrazione iniziale.
Sia ad esempio HA un acido debole generico, ka la sua costante di dissociazione e Ciniz la sua concentrazione iniziale. Se a è il suo grado di dissociazione, all'equilibrio si formeranno Ciniz mol/l di ioni H+ e Ciniz mol/l di ioni A-, mentre rimarranno indissociate (Ciniz - Ciniz ) mol/L di HA.
Riportiamo quanto detto in una tabella
Specie |
Concentrazioni iniziali |
Concentrazioni |
HA |
Ciniz |
Ciniz - α Ciniz |
H+ |
0 |
α Ciniz |
A- |
0 |
α Ciniz |
Se riportiamo ora i valori di equilibrio in funzione di ka, otteniamo

Da tale relazione (legge di diluizione di Ostwald) si deduce facilmente che, essendo ka costante, al diminuire della concentrazione iniziale il grado di dissociazione a deve aumentare. In altre parole anche un acido debole, se molto diluito, può essere quasi completamente dissociato. Tutti gli elettroliti tendono a dissociarsi completamente quando la concentrazione tende a zero.
Naturalmente il fatto che un acido debole a concentrazioni molto basse sia molto dissociato non significa che in tali condizioni diventi forte. Infatti a basse concentrazioni dell'acido anche gli ioni H+ che si producono sono complessivamente molto pochi ed il pH rimane sempre molto vicino a 7.
Calcoliamo ad esempio il pH ed il grado di dissociazione di una soluzione 1M e di una soluzione 10-2 M di acido fluoridrico (ka = 3,53.10-4).
L'acido fluoridrico è un acido debole e si dissocia secondo il seguente equilibrio
HF  H+ + F –
H+ + F –
Se poniamo pari ad x il numero di mol/l di HF che si dissociano, possiamo costruire la seguente tabella delle concentrazioni iniziali e delle concentrazioni di equilibrio
Specie |
Concentrazioni iniziali |
Concentrazioni |
HF |
1 |
1 - x |
H+ |
0 |
x |
F- |
0 |
x |
Poniamo ora le concentrazioni di equilibrio, espresse in funzione di x, in relazione con la ka

Risolvendo rispetto ad x si ottiene
[H+] = x = 1,86.10-2 mol/l
il pH vale
pH = -log [H+] = -log 1,86.10-2 = 1,73
mentre il grado di dissociazione risulta pari a

Risultano quindi dissociate quasi 2 molecole ogni 100.
Vediamo ora come varia il pH ed il grado di dissociazione diluendo la soluzione.
Se la concentrazione iniziale della soluzione è ora pari a 10-2 mol/L, la relazione di equilibrio diventa

Risolvendo rispetto ad x si ottiene
[H+] = x = 1,71.10-3 mol/l
il pH vale
pH = -log [H+] = -log 1,71.10-3 = 2,77
mentre il grado di dissociazione risulta pari a

Risultano quindi dissociate quasi 2 molecole ogni 10.
Diminuendo la concentrazione iniziale è dunque aumentata la percentuale di molecole che si dissociano. Nonostante questo il pH è aumentato, a dimostrazione del fatto che la seconda soluzione è meno acida della prima.
In generale per calcolare il grado di dissociazione è necessario risolvere la seguente equazione di secondo grado, che si ottiene riordinando la relazione di Ostwald

dove si osserva che il grado di dissociazione dipende dal rapporto tra la concentrazione iniziale dell’acido e la sua costante di dissociazione. Riportiamo a titolo di esempio alcuni valori del grado di dissociazione in funzione del rapporto C/ka.
C/ka |
106 |
105 |
104 |
103 |
102 |
101 |
100 |
10-1 |
10-2 |
10-3 |
α (%) |
0.1 |
0.3 |
1 |
3 |
9 |
27 |
62 |
92 |
99 |
99.9 |
Si noti che se C/ka > 102 il grado di dissociazione è inferiore al 10 %. In questa condizione la quantità di acido che si dissocia può essere consideratata trascurabile rispetto alla quantità di acido iniziale.
Inoltre, poichè per C/ka > 102 il grado di dissociazione α è molto inferiore all’unità, si avrà che (1 – α) ≈ 1 e la relazione di Ostwald può essere approssimata nel modo seguente

Se ne deduce che, per C/ka > 102 il grado di dissociazione α può essere stimato con la seguente relazione approssimata

Anche nel caso degli acidi deboli, come abbiamo visto per gli acidi forti, possiamo evitare di tener conto dell’equilibrio dall’acqua nel calcolo del pH solo se l’acido produce una quantità di ioni H+ sufficientemente elevata, da rendere trascurabili gli ioni H+ generati dall’acqua . In generale gli ioni H+ generati dall’acido dovrebbero almeno essere in quantità superiore od uguale a 10-6 mol/L ([H+]acido ≥ 10-6 M), in modo da poter trascurare i 10-7 mol/L generati dalla dissociazione dell’acqua
Nel caso degli acidi forti era sufficiente che la concentrazione dell’acido fosse superiore a 10-6 mol/L. Ma nel caso di un acido debole non è possibile far riferimento solo alla sua concentrazione iniziale C poiché la quantità di ioni H+ generati dipende anche dalla sua ka.
In generale è possibile trascurare l’equilibrio dell’acqua quando il prodotto tra la concentrazione iniziale e la costante di dissociazione dell’acido debole è superiore o uguale a 10-12 ed il rapporto tra la sua concentrazione iniziale e la sua costante di dissociazione è superiore o uguale a 102.
C·ka ≥ 10-12  ≥ 102
≥ 102
Dunque, se entrambe le condizioni sono soddisfatte, possiamo trascurare gli ioni H+ provenienti dalla dissociazione dell’acqua e calcolare il pH considerando solo l’equilibrio dell’acido debole, risolvendo la seguente equazione di secondo grado


con x = [H+]
Come vedremo, se tenessimo conto anche degli ion H+ provenienti dall’acqua, dovremmo risolvere un’equazione di terzo grado.
In realtà le condizioni poste per poter trascurare l’equilibrio dell’acqua ci permettono di evitare di risolvere l’equazione di secondo grado, utilizzando un metodo risolutivo semplificato.
Metodo semplificato per il calcolo del pH di acidi e basi deboli
Consideriamo il solito acido debole generico HA. L'equilibrio di dissociazione è
HA  H+ + A–
H+ + A–
e la costante di equilibrio vale

Se poniamo pari ad x il numero di mol/L di HA che si dissociano, possiamo costruire la seguente tabella delle concentrazioni iniziali e delle concentrazioni di equilibrio
Specie |
Concentrazioni iniziali |
Concentrazioni |
HA |
C |
C - x |
H+ |
0 |
x |
A- |
0 |
x |
Poniamo ora, come al solito, le concentrazioni di equilibrio, espresse in funzione di x, in relazione con la ka

Abbiamo visto nel paragrafo precedente che, nel caso in cui C·ka ≥ 10-12 e  ≥ 102 l’acido risulta anche poco dissociato, presentando un grado di dissociazione α < 0.1.
≥ 102 l’acido risulta anche poco dissociato, presentando un grado di dissociazione α < 0.1.
Questo significa che la quantità x di acido che si dissocia è piccola e trascurabile rispetto alla concentrazione iniziale C dell’acido (x << C). Possiamo allora ragionevolmente assumere che all’equilibrio la quantità (C – x) sia praticamente uguale a C
C – x ≈ C
Trascurando dunque la x nella differenza a denominatore nella equazione di equilibrio, otterremo


che ci permette di calcolare la concentrazione tramite la seguente relazione semplificata

Eseguendo il logaritmo decimale negativo di entrambi i membri si ha
pH = ½ (pka –logC)
e, per una base debole,

pOH = ½ (pkb –logC)
Nella tabella seguente riportiamo il valore del pH calcolato per l’intervallo di valori di C e ka all’interno del quale è possibile usare il metodo approssimato (C·ka ≥ 10-12 e  ≥ 102).
≥ 102).
In ogni casella compaiono 3 valori di pH calcolati rispettivamente
1) con l’equazione di terzo grado che tiene conto anche della dissociazione dell’acqua (valore esatto)
2) con l’equazione di secondo grado che trascura la dissociazione dell’acqua
3) con la relazione semplificata che trascura la x a denominatore nell’equazione di secondo grado
pH |
Costante di dissociazione acida - Ka |
|||||||||||
C mol/L |
|
10-2 |
10-3 |
10-4 |
10-5 |
10-6 |
10-7 |
10-8 |
10-9 |
10-10 |
10-11 |
10-12 |
1 |
1,02 |
1.51 |
2.00 |
2.50 |
3.00 |
3.50 |
4.00 |
4.50 |
5.00 |
5.50 |
6.00 |
|
10-1 |
|
2.02 |
2.51 |
3.00 |
3.50 |
4.00 |
4.50 |
5.00 |
5.50 |
6.00 |
|
|
10-2 |
|
|
3.02 |
3.51 |
4.00 |
4.50 |
5.00 |
5.50 |
6.00 |
|
|
|
10-3 |
|
|
|
4.02 |
4.51 |
5.00 |
5.50 |
6.00 |
|
|
|
|
10-4 |
|
|
|
|
5.02 |
5.51 |
6.00 |
|
|
|
|
|
10-5 |
|
|
|
|
|
6.02 |
|
|
|
|
|
|
Si noti come la relazione semplificata fornisca valori migliori quando il rapporto C/ka è più elevato e quindi l’acido è meno dissociato (grado di dissociazione basso e quindi x molto piccola rispetto a C).
Calcolo del pH di acidi e basi deboli molto diluiti e/o molto deboli.
Quando un acido debole è molto diluito e/o molto debole non è possibile trascurare, nel calcolo del pH, la concentrazione degli ioni H+ provenienti dalla dissociazione dell’acqua.
L’equilibrio dell’acido e dell’acqua si disturbano reciprocamente.
La quantità di acido che si dissocia è infatti inferiore rispetto a quel che avremo calcolato senza tener conto dell’acqua, a causa degli ioni H+ prodotti dall’acqua che spostano verso sinistra l’equilibrio dell’acido.
In modo analogo l’acqua si dissocia meno per la presenza degli ioni H+ generati dall’acido.
Il pH si calcola sommando gli ioni H+ generati dall’acqua agli ioni H+ generati dall’acido.
Indichiamo allora con x la concentrazione di ioni H+ generati dall’acido e con y la concentrazione di ioni H+ generati dall’acqua. La concentrazione totale di ioni H+ sarà (x + y) e tale quantità dovrà soddisfare contemporaneamente l’equilibrio dell’acido e l’equilibrio dell’acqua.
Ora consideriamo un generico acido debole HA di concentrazione iniziale C e costante di dissociazione acida Ka. L’acido si dissocia, in presenza di y ioni H+ dell’acqua, in x ioni H+ ed x ioni A–.
Scriviamo dunque l’equilibrio dell’acido riportando sotto ogni specie chimica le concentrazioni di equilibrio
HA → H+ + A–
C – x x + y x
Consideriamo ora l’equilibrio dell’acqua che si dissocia, in presenza degli x ioni H+ provenienti dalla dissociazione dell’acido, in y ioni H+ ed y ioni OH-. Scriviamo dunque l’equilibrio dell’acqua riportando sotto ogni specie chimica le concentrazioni di equilibrio
H2O → H+ + OH–
55,55 - y x + y y
Scriviamo ora le rispettive relazioni di equilibrio, sostituendo opportunamente le concentrazioni di equilibrio
1) 
2) 
Dalla relazione 2) ricaviamo il valore della  che sostituiamo nella 1) ottenendo la seguente equazione di terzo grado in y
che sostituiamo nella 1) ottenendo la seguente equazione di terzo grado in y
3) 
Calcoliamo ad esempio il pH di una soluzione 0.5 M di un acido debole con ka = 10-14.
l’equazione 3) fornisce y = [H+]acqua = 8,1650 10-8 M
Si noti che l’acqua in assenza dell’acido produce una concentrazione di ioni H+ pari a 10-7 mol/L, mentre qui, a causa della presenza dell’acido, ne produce solo 8,1650 10-8 mol/L
Ora usiamo il valore trovato della y per sostituirlo nella 2) e calcolare il valore della x (concentrazione di ioni H+ generata dall’acido)

da cui
x = [H+]acido = 4,0825 ·10-8 M
Si noti che se avessimo calcolato la concentrazione di ioni H+ generata dall’acido senza considerare l’acqua, utilizzando l’equazione di secondo grado, avremo trovato un valore superiore, pari a


x = [H+] = 7,0711 ·10-8 M
L’acido si dissocia quindi meno di quanto farebbe in assenza degli ioni H+ generati dall’acqua. Tuttavia se considerassimo solo gli ioni H+ dell’acido commetteremmo in questo caso un errore. Si noti infatti come la concentrazione di ioni H+ prodotta dall’acido sia dello stesso ordine di grandezza di quella proveniente dall’acqua. E dunque quest’ultima non possa essere trascurata.
La concentrazione totale di ioni H+ è quindi pari a
y + x = [H+]acqua + [H+]acido = 8,1650 10-8 + 4,0825 10-8 = 1.225 10-7 M
che porta ad un pH = 6.91
Se avessimo considerato solo l’equilibrio dell’acido saremmo arrivati al risultato palesemente assurdo di un pH basico (pH = - log 7,0711 ·10-8 = 7.15)
L’equazione 3) ci permette di calcolare in modo esatto la concentrazione di ioni H+ generata dall’acido debole e di sommarla successivamente agli ioni H+ prodotti dall’acqua per ottenere la concentrazione totale degli ioni H+.
E’ tuttavia possibile ricavare un’equazione che fornisca direttamente la concentrazione totale degli ioni H+. Per trovarla riconsideriamo i due equilibri che dobbiamo analizzare e che si disturbano reciprocamente, quello dell’acido debole e quello dell’acqua.
HA → H + + A–
H2O → H + + OH –
Nei due equilibri compaiono le seguenti 4 incognite.
1) [H+] 2) [OH –] 3) [HA] 4) [A–]
Dobbiamo pertanto scrivere 4 equazioni indipendenti nelle 4 incognite.
La prima e la seconda equazione si ricavano dalle relazioni di equilibrio rispettivamente dell’acido e dell’acqua
a) 
b) 
la terza si ottiene dal bilancio delle cariche (la somma di tutte le cariche positive deve essere uguale alla somma di tutte le cariche negative)
c) 
la quarta si ricava infine dal bilancio di massa (il numero iniziale C di molecole dell’acido deve essere uguale alla somma delle molecole di acido indissociato HA e delle molecole di acido dissociato A– all’equilibrio)
d) 
Ricaviamo ora  dalla relazione d) e sostituiamo nella relazione a)
dalla relazione d) e sostituiamo nella relazione a)
e) 
Ricaviamo  dalla relazione c) e sostituiamo nella e)
dalla relazione c) e sostituiamo nella e)
f) 
Si ricava infine  dalla relazione b) e si sostituisce nella f) che, riordinata, fornisce
dalla relazione b) e si sostituisce nella f) che, riordinata, fornisce
4) 
un’equazione di terzo grado che ci permette di calcolare il valore esatto della concentrazione totale di ioni H+ per una soluzione qualsiasi di un acido debole.
È evidente che risolvere un’equazione di terzo grado non è affatto una prospettiva allettante. Vediamo allora se è possibile sostituirla con metodi risolutivi più semplici, anche se, ovviamente, approssimati. Consideriamo i seguenti tre casi
Caso 1) Acidi debolissimi Ka ≤ 10-7 (e C·Ka < 10-12 )
Consideriamo l’equazione risolutiva esatta di terzo grado (equazione 4) e dividiamola per la concentrazione degli ioni H+, ottenendo
5) 
Verifichiamo ora come, nelle condizioni considerate (Ka ≤ 10-7), i termini  risultino entrambi più piccoli e quindi trascurabile rispetto al termine Kw.
risultino entrambi più piccoli e quindi trascurabile rispetto al termine Kw.
Infatti, essendo la soluzione acida, si avrà [H+] > 10-7 ed essendo Ka ≤ 10-7 quindi  dunque
dunque

Inoltre, essendo C·Ka ≤ 10-12, la concentrazione degli ioni H+ deve essere non molto diversa da 10-7 per cui
 ≈ ka 10-7 < 10-14
≈ ka 10-7 < 10-14
che è ciò che volevamo verificare.
Possiamo dunque trascurare i termini  e l’equazione 5) può quindi essere ridotta di grado. diventando
e l’equazione 5) può quindi essere ridotta di grado. diventando

e la concentrazione degli ioni H+ è calcolabile con la seguente formula semplificata
6) 
Proviamo ad applicare tale formula risolutiva all’esempio precedente: una soluzione 0.5 M di un acido debole con Ka = 10-14, che avevamo risolto utilizzando l’equazione di terzo grado ottenendo
[H+] = 1.225 10-7 M e pH = 6.91
In questo caso il prodotto C·Ka = 0.5 10-14 è inferiore a 10-12 Ciò significa che gli ioni H+ generati dall’acqua non sono trascurabili rispetto a quelli prodotti dall’acido e non possiamo pertanto utilizzare la relazione semplificata  (la quale fornirebbe [H+] = 7.07 10-8 M ed un pH = 7.15, basico!!).
(la quale fornirebbe [H+] = 7.07 10-8 M ed un pH = 7.15, basico!!).
Applicando invece la formula 6) otteniamo
 1.225 10-7 M e pH = 6.91
1.225 10-7 M e pH = 6.91
Il medesimo risultato ottenuto con l’equazione di terzo grado!!! Non male.
Caso 2) Acidi deboli molto diluiti Ka > 10-7 e C ≤ 10-7
Per acidi così diluiti il grado di dissociazione risulta molto elevato. Se Ka > 10-7 e C ≤ 10-7 il rapporto C/ka risulta essere infatti inferiore all’unità ed il grado di dissociazione superiore al 70%. In queste condizioni l’acido, pur rimanendo un acido debole, può essere trattato come un acido forte completamente dissociato. Possiamo cioè calcolare come si comporta l’equilibrio dell’acqua in presenza di C ioni H+ prodotti dall’acido debole completamente dissociato
HA → H+ + A–
C – C + C + C
H2O → H+ + OH–
55,55 – x x + C x
Kw = [H+]·[OH–] = (x + C) x
x2 + Cx – Kw = 0
e la concentrazione degli ioni H+ è calcolabile con la seguente formula

3) Acidi deboli che non ricadono nei casi precedenti (C > 10-7 e C/·ka < 102 )
Consideriamo il caso di un acido debole che non ricada nei casi precedenti (per i quali abbiamo già individuato una formula risolutiva semplificata).
Consideriamo ancora l’equazione risolutiva esatta di terzo grado (equazione 4) divisa per la concentrazione degli ioni H+

è possibile verificare come, nelle condizioni considerate il termine  risulti più piccolo e quindi trascurabile rispetto al termine kaC.
risulti più piccolo e quindi trascurabile rispetto al termine kaC.
Essendo infatti la soluzione acida, si avrà [H+] > 10-7 , quindi 
Dunque, poiché per ipotesi C > 10-7 , allora  . Moltiplicando entrambi i membri della disuguaglianza per ka, otteniamo
. Moltiplicando entrambi i membri della disuguaglianza per ka, otteniamo

che è quanto volevamo verificare
L’equazione può quindi essere ridotta di grado. diventando

e la concentrazione degli ioni H+ è calcolabile con la seguente formula

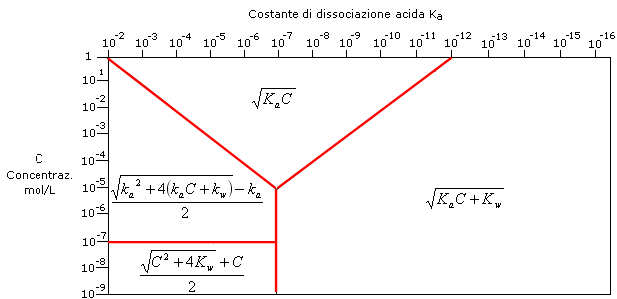
Possiamo dunque utilizzare 4 formule risolutive approssimate che si applicano in condizioni diverse di concentrazione (C) e di forza (ka) dell’acido.
Nello schema seguente vengono riportate le 4 formule risolutive per il calcolo della concentrazione degli ioni H+ in funzione di C e ka.
Riportiamo infine una tabella con alcuni valori di pH calcolati con le relazioni semplificate, confrontati con i valori esatti. In ogni casella è presente il vaore esatto (in nero sopra) ed il valore approssimato (in colore sotto)
pH |
Costante di dissociazione acida - Ka |
||||||||||||||||
C mol/L |
|
10-2 |
10-3 |
10-4 |
10-5 |
10-6 |
10-7 |
10-8 |
10-9 |
10-10 |
10-11 |
10-12 |
10-13 |
10-14 |
10-15 |
10-16 |
|
1 |
1,02 |
1.51 |
2.00 |
2.50 |
3.00 |
3.50 |
4.00 |
4.50 |
5.00 |
5.50 |
6.00 |
6.48 |
6.85 |
6.98 |
7.00 |
|
|
10-1 |
1.57 |
2.02 |
2.51 |
3.00 |
3.50 |
4.00 |
4.50 |
5.00 |
5.50 |
6.00 |
6.48 |
6.85 |
6.98 |
7.00 |
7.00 |
|
|
10-2 |
2.21 |
2.57 |
3.02 |
3.51 |
4.00 |
4.50 |
5.00 |
5.50 |
6.00 |
6.48 |
6.85 |
6.98 |
7.00 |
7.00 |
7.00 |
|
|
10-3 |
3.04 |
3.21 |
3.57 |
4.02 |
4.51 |
5.00 |
5.50 |
6.00 |
6.48 |
6.85 |
6.98 |
7.00 |
7.00 |
7.00 |
7.00 |
|
|
10-4 |
4.00 |
4.04 |
4.21 |
4.57 |
5.02 |
5.51 |
6.00 |
6.48 |
6.85 |
6.98 |
7.00 |
7.00 |
7.00 |
7.00 |
7.00 |
|
|
10-5 |
5.00 |
5.00 |
5.04 |
5.21 |
5.57 |
6.02 |
6.49 |
6.85 |
6.98 |
7.00 |
7.00 |
7.00 |
7.00 |
7.00 |
7.00 |
|
|
10-6 |
6.00 |
6.00 |
6.00 |
6.03 |
6.20 |
6.54 |
6.86 |
6.98 |
7.00 |
7.00 |
7.00 |
7.00 |
7.00 |
7.00 |
7.00 |
|
|
10-7 |
6.79 |
6.79 |
6.79 |
6.79 |
6.82 |
6.90 |
6.98 |
7.00 |
7.00 |
7.00 |
7.00 |
7.00 |
7.00 |
7.00 |
7.00 |
|
|
10-8 |
6.98 |
6.98 |
6.98 |
6.98 |
6.98 |
6.99 |
7.00 |
7.00 |
7.00 |
7.00 |
7.00 |
7.00 |
7.00 |
7.00 |
7.00 |
|
|
10-9 |
7.00 |
7.00 |
7.00 |
7.00 |
7.00 |
7.00 |
7.00 |
7.00 |
7.00 |
7.00 |
7.00 |
7.00 |
7.00 |
7.00 |
7.00 |
|
|
Riassumendo
Per calcolare la concentrazione degli ioni H+ di una soluzione di un acido debole monoprotico è possibile utilizzare una delle quattro seguenti formule semplificate in relazione ai valori che assumono la concentrazione C e la costante di dissociazione acida ka.
1) Se C·Ka ≥ 10-12 e  ≥ 102 si utilizza la relazione
≥ 102 si utilizza la relazione

2) Se Ka ≤ 10-7 e C·Ka < 10-12 si utilizza la relazione

3) Se C ≤ 10-7 e Ka > 10-7 si utilizza la relazione

4) Se C > 10-7 e  < 102 si utilizza la relazione
< 102 si utilizza la relazione

pH in soluzioni di acidi e basi deboli poliprotici
Il calcolo del pH di acidi e basi deboli poliprotici, implicando più equilibri di dissociazione, ognuno caratterizzato da una propria costante di equilibrio, risulta essere più complesso. In generale è infatti necessario risolvere sistemi di più equazioni.
Molto spesso accade però che le costanti di dissociazione successive alla prima presentino valori molto minori. In tal caso è possibile dimostrare che la concentrazione di equilibrio degli ioni H+ dipende in pratica solo dal primo equilibrio. Inoltre i calcoli per la determinazione di tutte le concentrazioni di equilibrio possono venire notevolmente semplificati considerando ciascun equilibrio di dissociazione separatamente ed in modo indipendente dagli altri.
Per esemplificare quanto affermato calcoliamo il pH di una soluzione 0,5 M di acido solforoso, sapendo che la costante di prima dissociazione è  .
.
I due equilibri di dissociazione sono
H2SO3  H+ + HSO3-
H+ + HSO3-
HSO3-  H+ + SO32-
H+ + SO32-
Per risolvere correttamente il problema è necessario considerare contemporaneamente i due equilibri in quanto gli ioni H+ prodotti dalla prima dissociazione tendono a spostare verso sinistra il secondo equilibrio e viceversa.
Chiamando x il numero di mol/L di acido solforoso che si dissociano nel primo equilibrio e y il numero di mol/L di HSO3- che si dissociano nel secondo equilibrio, avremo
per il primo equilibrio
Specie |
Concentrazioni iniziali |
Concentrazioni |
H2SO3 |
0,5 |
0,5 - x |
H+ |
0 |
x + y |
HSO3- |
0 |
x - y |
Infatti alle x mol/L di ioni H+ prodotti dal primo equilibrio è necessario aggiungere le y mol/L di ioni H+ prodotti dal secondo equilibrio, mentre alle x mol/L di anioni HSO3- prodotti dal primo equilibrio è necessario togliere le y mol/L che si dissociano nel secondo equilibrio.
per il secondo equilibrio
Specie |
Concentrazioni iniziali |
Concentrazioni |
HSO3- |
0 |
x - y |
H+ |
0 |
x + y |
SO32- |
0 |
y |
Sarebbe quindi necessario risolvere il seguente sistema di equazioni, che garantisce che entrambe le condizioni di equilibrio siano contemporaneamente soddisfatte.


La risoluzione risulta però lunga e laboriosa, generando tra l'altro un'equazione di grado superiore al secondo.
Nel caso si tenga conto anche dell’equilibrio di dissociazione dell’acqua la formula risolutiva esatta è

con x = [H+]
mentre, se si trascura l’equilibrio di dissociazione dell’acqua, l’equazione si abbassa di un grado e si ha

In questo caso possiamo comunque risolvere il problema in modo approssimato poiché la costante di seconda dissociazione risulta essere di ben 5 ordini di grandezza inferiore della costante di prima dissociazione e possiamo quindi ragionevolmente ritenere che gli ioni H+ prodotti dal secondo equilibrio siano trascurabili rispetto a quelli prodotti dal primo. E' possibile quindi considerare il primo equilibrio di dissociazione prevalente e procedere alla soluzione separata dei due equilibri.
Prendiamo dunque in considerazione il primo equilibrio come se non fosse presente il secondo

La soluzione dell'equazione di 2° grado ci fornisce il seguente valore
x = [H+]I = [HSO3-] = 6,59.10-2
Dove [H+]I rappresenta la concentrazione di ioni H+ prodotti dalla prima dissociazione.
Utilizziamo ora la concentrazione di HSO3- trovata, come concentrazione iniziale per la seconda dissociazione e teniamo conto in questo caso che gli ioni H+ provenienti dalla prima dissociazione spostano l'equilibrio verso sinistra

Essendo la ka estremamente piccola y avrà un valore che potrà essere tranquillamente trascurato sia nella somma a numeratore che nella differenza a denominatore. Otteniamo in tal modo il seguente risultato
y = [H+]II = [SO32-] = 2,6.10-7
Come si può notare la concentrazione di ioni H+ provenienti dalla seconda dissociazione è talmente bassa che, anche se sommata alla concentrazione degli ioni H+ proveniente dalla prima dissociazione non ne modifica il valore
[H+]tot = [H+]I + [H+] II = x + y = 6,59.10-2 + 2,6.10-7 = 6,59.10-2
Possiamo inoltre verificare che gli ioni H+ provenienti dalla seconda dissociazione sono in concentrazione talmente esigua da giustificare la trattazione separata del primo equilibrio. La loro presenza in soluzione sposta infatti l'equilibrio di prima dissociazione verso sinistra di una quantità assolutamente trascurabile.
In generale è possibile trattare gli equilibri separatamente, senza commettere grossi errori, quando le costanti di dissociazione differiscono di almeno 3 - 4 ordini di grandezza.
Naturalmente quando si considerano separatamente i due equilibri valgono tutte le considerazioni fatte per gli acidi deboli monoprotici.
Sarà dunque necessario scegliere la formula risolutiva semplificata più appropriata in relazione ai valori della concentrazione C e della costante di prima dissociazione acida ka1.
Esempio
Calcoliamo [H+] di una soluzione 10-1 M di acido metasilicico H2SiO3 (ka1 = 2 10-10, ka2 =1 10-12) e di una soluzione 10-5 M del medesimo acido.
Consideriamo solo l’equilibrio di prima dissociazione e verifichiamo le condizioni di applicabilità delle formule semplificate per gli acidi deboli monoprotici.
- Per la soluzione 10-1 M il prodotto C·Ka1 = 2 10-11 > 10-12 ed il rapporto
 = 5 108 > 102 . Possiamo dunque applicare la prima formula semplificata
= 5 108 > 102 . Possiamo dunque applicare la prima formula semplificata

- Per la soluzione 10-5 M il prodotto C·Ka1 = 2 10-15 < 10-12 non possia mo dunque usare la prima formula semplificata
 . Tuttavia, poiché ka1 = 10-12 < 10-7 possiamo usare la seconda formula semplificata
. Tuttavia, poiché ka1 = 10-12 < 10-7 possiamo usare la seconda formula semplificata

Indicatori di pH
Il pH di una soluzione viene normalmente misurato tramite uno strumento detto piaccametro. Per ottenere una indicazione qualitativa del pH di una soluzione si usano particolari sostanze chimiche dette indicatori.
Un indicatore è generalmente costituito da un acido o da una base debole che assume un colore diverso in relazione al pH della soluzione.
Il valore del pH in corrispondenza del quale un indicatore cambia colore è detto pH di viraggio.
Usando un indicatore unico non è quindi possibile sapere qual è il pH di una soluzione, ma solo se la soluzione presenta un pH superiore o inferiore al pH di viraggio di quell'indicatore.
Per conoscere il valore approssimato del pH di una soluzione è necessario utilizzare una striscia di carta impregnata con più indicatori (indicatore universale), che assume diversi colori a seconda del pH.
Se indichiamo con HIn un generico indicatore costituito da un acido debole, il suo equilibrio di dissociazione sarà
HIn  H+ + In-
H+ + In-
La capacità dell'indicatore di assumere due diversi colori è legata al fatto che la forma indissociata HIn presenta un colore diverso dell'anione In-.
L'equilibrio dell'indicatore si sposta per il principio di Le Chatelier verso destra o verso sinistra, facendo prevalere una delle due forme colorate sull'altra, a seconda che esso si trovi in presenza di una soluzione basica o acida.
Infatti se aumentiamo la [H+] l'equilibrio dell'indicatore tende a spostarsi verso sinistra e prevale HIn, mentre se diminuiamo la [H+] l'equilibrio si sposta verso destra e prevale In-.
Così se l'indicatore viene posto in una soluzione acida, parte degli ioni H+ presenti in soluzione reagiscono con gli ioni In- per ridare la forma indissociata HIn e l'equilibrio dell'indicatore si sposta verso sinistra.
Mentre se l'indicatore viene posto in una soluzione basica, parte degli ioni OH- presenti in soluzione si associano con gli ioni H+ provenienti dalla dissociazione dell'indicatore per formare H2O indissociata (l'equilibrio di dissociazione dell'acqua è infatti molto spostato verso sinistra) . La diminuzione degli ioni H+ costringe l'indicatore a dissociarsi ulteriormente ed il suo equilibrio si sposta verso destra.
Naturalmente il punto di viraggio viene raggiunto quando la concentrazione della specie indissociata HIn eguaglia la concentrazione dell'anione In-.
punto di viraggio [HIn] = [In-]
Il pH di viraggio è facilmente determinabile conoscendo il valore della costante di dissociazione dell'indicatore, infatti

da cui

ricordando infine che al punto di viraggio [HIn] = [In-],
allora 
e quindi

passando infine ai logaritmi, si dimostra che il pH di viraggio è uguale al logaritmo negativo della costante di dissociazione dell'indicatore o pK

Naturalmente se l’indicatore è una base debole sarà

e

In realtà ciascun indicatore vira (cambia colore) in corrispondenza di un intervallo di pH necessario affinché l'occhio possa apprezzare il cambiamento di colore. L’occhio umano riesce infatti ad apprezzare il colore quando le due concentrazioni variano almeno di 10 volte.
Ad esempio per un generico indicatore acido HIn, il colore A predomina quando il rapporto [In–]/[HIn] < 1/10 ed il colore B quando [In–]/[HIn] > 10
L’intervallo di pH di viraggio di un indicatore sarà dunque pari a pkind ± 1. Supponendo ad esempio di impiegare un indicatore acido con ka= 10-5, il viraggio del colore verrà apprezzato quando il pH varia tra 6 e 4 (intervallo di viraggio dell’indicatore). Infatti
- quando [HIn] = 10 [In–] si avrà

- quando [In–] = 10 [HIn] si avrà

Indicatore |
Intervallo di |
Colore |
|
Forma acida |
Forma basica |
||
Blu timolo |
1,2 – 2,8 |
Rosso |
Giallo |
|
8,0 – 9,6 |
Giallo |
Blu |
Blu bromofenolo |
3,0 – 4,6 |
Giallo |
Blu |
Metilarancio |
3,1 – 4,4 |
Rosso |
Giallo-rame |
Rosso congo |
3,0 – 5,0 |
Blu |
Rosso |
Verde di bromocresolo |
3,9 – 5,6 |
Giallo |
Blu |
Rosso di metile |
4,4 – 6,3 |
Rosso |
Giallo |
Bromocresolo porpora |
5,2 – 6,8 |
Giallo |
Porpora |
Blu di bromotimolo |
6,2 – 7,6 |
Giallo |
Blu |
Rosso fenolo |
6,4 – 8,0 |
Giallo-arancio |
Rosso |
Cresolo porpora |
7,6 – 9,2 |
Giallo |
Rosso-porpora |
Fenolftaleina |
8,0 – 9,9 |
Incolore |
Rosso |
Timoftaleina |
9,3 – 10,6 |
Incolore |
Blu |
Giallo di alizarina |
10,1 – 12,0 |
Incolore |
Violetto |
Idrolisi salina ed equilibrio di idrolisi
Non tutte le soluzioni saline sono neutre. Si osserva infatti sperimentalmente che alcune soluzioni saline sono neutre, alcune basiche ed altre ancora acide.
Tale fenomeno è legato alla possibilità che alcuni ioni provenienti dalla dissociazione del sale reagiscano con l'acqua per ridare parzialmente l'acido o la base da cui è derivato il sale.
Tale reazione è detta di idrolisi salina e per questo motivo a volte l'idrolisi salina viene considerata come una reazione inversa della reazione di salificazione.
Naturalmente si distingue un'idrolisi neutra, un'idrolisi basica ed un'idrolisi acida in relazione al pH della soluzione salina.
Idrolisi basica
(sale derivato da base forte ed acido debole)
Si produce un'idrolisi basica quando viene sciolto in acqua un sale il cui anione deriva da un acido debole, mentre il catione metallico deriva da una base forte.
Prendiamo ad esempio l'ipoclorito di sodio NaClO, che deriva dall'acido ipocloroso HClO (acido debole con ka piccola) e dall'idrossido di sodio NaOH (base forte con kb elevata).
In soluzione acquosa l'ipoclorito è, come la maggior parte dei sali, completamente dissociato in ioni Na+ e ioni ClO-
NaClO  Na+ + ClO-
Na+ + ClO-
E' necessario ora analizzare in che modo tali ioni interferiscano con l'equilibrio di dissociazione dell'acqua
H2O  H+ + OH-
H+ + OH-
Ora, mentre lo ione Na+ non ha alcuna tendenza a riassociarsi con l'anione OH- per dare l'idrossido di sodio indissociato, poiché NaOH è una base forte ed il suo equilibrio di dissociazione è completamente spostato verso destra;
NaOH → Na+ + OH-
l'anione ClO- presenta una grande tendenza a riassociarsi con gli ioni H+ prodotti dalla dissociazione dell'acqua per ridare l'acido ipocloroso, il cui equilibrio di dissociazione è invece fortemente spostato verso sinistra
H+ + ClO- → HClO
Poiché dunque lo ione ipoclorito ruba ioni H+ all'equilibrio di dissociazione dell'acqua, quest'ultima, per il principio di Le Chatelier, sposta il suo equilibrio verso destra producendo altri ioni H+ e naturalmente altrettanti ioni OH-. Naturalmente all'equilibrio, poiché gli ioni H+ vengono assorbiti dall'acido ipocloroso che si riassocia, in soluzione rimarrà un eccesso di ioni OH-.
Per calcolare il pH di tali soluzioni sarebbe necessario considerare congiuntamente i due equilibri che interferiscono, quello dell'acqua e quello dell'acido debole, in modo da soddisfare contemporaneamente le relative equazioni di equilibrio.
Se indichiamo con
y = la quantità di acqua che si dissocia liberando y mol/L di ioni OH- e y mol/L di ioni H+
x = la quantità di anione ipoclorito che si riassocia rubando x mol/L di ioni H+ per formare x mol/L di acido indissociato
C = concentrazione iniziale del sale = concentrazione iniziale dell'anione ipoclorito


Le due equazioni formano un sistema che richiede la soluzione di un'equazione di grado superiore al secondo.
Soluzione esatta Per calcolare il pH di tale soluzione sarebbe necessario tener conto simultaneamente dei seguenti due equilibri 1) H2O 2) HClO Nei 2 equilibri compaiono le seguenti 4 incognite. 1) [H+] 2) [OH–] 3) [HClO] 4) [ClO–]
Dobbiamo pertanto scrivere 4 equazioni indipendenti nelle 4 incognite.
la terza si ottiene dal bilancio delle cariche (la somma di tutte le cariche positive deve essere uguale alla somma di tutte le cariche negative)
la quale, ricordando che il sale è completamente dissociato e quindi [Na+] = C, diventa
la quarta si ricava infine dal bilancio di massa. Ricordando che il sale è completamente dissociato in C mol/L di ipoclorito ClO– e C mol/L di ioni sodio Na+, dopo l’idrolisi la somma dell’acido indissociato che si forma e dell’anione residuo all’equilibrio dovrà essere pari alla concentrazione iniziale C e potremo quindi scrivere
utilizzando le 4 equazioni ed effettuando le opportune sostituzioni si ottiene
un’equazione di terzo grado che ci permette di calcolare il valore esatto della concentrazione totale di ioni H+ per una soluzione di un sale formato da un acido debole ed una base forte. |
 H+ + OH-
H+ + OH-  H+ + ClO-
H+ + ClO- 





Quando la concentrazione iniziale del sale è sufficientemente elevata, è possibile evitare di ricorrere alla soluzione esatta del problema, introducendo alcune semplificazioni nella trattazione.
Si ipotizza cioè che l'equilibrio dell'acido si sposti verso sinistra in misura pari allo spostamento verso destra dell'equilibrio dell'acqua. In altre parole per ogni molecola di acido che si forma dalla unione di un anione ClO- con un H+, una molecola d'acqua si dissoci per ridare lo ione H+ e uno ione OH-.
In questo modo si devono formare all'equilibrio tante molecole di HClO quanti ioni OH-.
In effetti ciò rappresenta solo una approssimazione in quanto in questo modo la concentrazione degli ioni H+ rimarrebbe inalterata, pari a 10-7, mentre la concentrazione degli ioni OH- crescerebbe ed il loro prodotto non soddisferebbe più la kw. In realtà parte degli ioni OH- si legano con gli ioni H+ in modo da soddisfare il prodotto ionico dell'acqua.
La concentrazione di equilibrio degli ioni OH- risulta pertanto leggermente inferiore di quella calcolata tenendo conto solamente della riassociazione dell'acido.
L'entità di tale processo è comunque minima e non influisce sulla concentrazione degli ioni OH- la quale è determinata essenzialmente dall'equilibrio dell'acido che si riassocia. Diviene necessario tener conto anche dell'equilibrio dell'acqua solo quando il sale è molto diluito.
La reazione semplificata che si ipotizza avvenga è detta reazione di idrolisi ed è la seguente
ClO- + H2O  HClO + OH-
HClO + OH-
E' facile verificare che la sua costante di equilibrio, detta costante di idrolisi o kh, vale

Tale relazione mette in evidenza come l'equilibrio di idrolisi basica è tanto più spostato verso destra quanto più l'acido è debole (maggiore tendenza ad associarsi liberando ioni OH-). Di conseguenza la soluzione risulterà essere tanto più basica quanto più piccolo è il valore della ka (e più elevato il valore della kh).
Per risolvere i problemi relativi agli equilibri di idrolisi si possono usare le stesse formule semplificate utilizzate per acidi e basi deboli monoprotici.
Esempio 1
Calcoliamo il pH di una soluzione 0,3 M di nitrito di potassio, sapendo che la ka dell'acido nitroso è pari 4,6.10-4 e di una soluzione 10-4 M del medesimo sale.
Il nitrito di potassio si dissocia completamente in 0,3 mol/L di ioni K+ e 0,3 mol/L di ioni NO2-.
KNO2 → K+ + NO2-
L'anione nitrito tende a riassociarsi con gli ioni H+ provenienti dall'acqua producendo il seguente equilibrio di idrolisi
NO2- + H2O  HNO2 + OH-
HNO2 + OH-
Calcoliamo la costante dell'equilibrio di idrolisi

Indichiamo con x la quantità di NO2- che si riassocia per dare x mol/L di HNO2, mentre vengono contemporaneamente liberate x mol/Ldi ioni OH-,
|
iniziale |
d'equilibrio |
[NO2-] |
0,3 |
0,3 - x |
[HNO2] |
0 |
x |
[OH-] |
0 |
x |
Esprimiamo le concentrazioni di equilibrio in funzione della costante di idrolisi

risolvendo l'equazione otteniamo
x = [OH-]= [HNO2] = 2,55.10-6 mol/L
[H+] = kw/ [OH-] = 3,92.10-9 mol/L pH = 8,4
(l’equazione di terzo grado fornisce [H+] = 3,91.10-9 mol/L)
Si noti come anche in questo caso era possibile ricorrere ad una soluzione semplificata in quanto la concentrazione iniziale del sale è sufficientemente elevata e la kh è sufficientemente piccola da far ritenere che la quantità di ioni H+ che si riassocia sia trascurabile rispetto alla concentrazione dell'anione.
Se dunque trascuriamo la x nella differenza a denominatore, possiamo usare la seguente relazione semplificata

dove Csale è la concentrazione iniziale del sale.
Il prodotto (kh∙C) vale 6.5 10-12 ed essendo maggiore di 10-12 ci permette di usare la prima formula semplificata per il calcolo del pH
Nel caso della soluzione 10-4 M invece, il prodotto kh∙C = 2.17 10-11 ∙ 10-4 = 6.5 10-15 < 10-12 e non possiamo quindi usare la formula semplificata  .
.
Poiché kh = 2.17 10-11 < 10-7 possiamo usare la seconda formula semplificata 

[H+] = kw/ [OH-] = 9,063.10-8 mol/L pH = 7,04
(l’equazione di terzo grado fornisce [H+] = 9,065.10-9 mol/L)
Esempio 2
Calcoliamo il pH di una soluzione 3 10-2 M di carbonato di sodio Na2CO3, sapendo che le costanti di prima e seconda dissociazione acida dell’acido carbonico valgono rispettivamente ka1 = 4,3 10-7 e ka2 = 5,6.10-11 e di una soluzione 10-4 M del medesimo sale.
Il carbonato di sodio si dissocia completamente in 6 10-2 di ioni Na+ e 3 10-2 mol/L di ioni CO32-
Na2CO3 → 2Na+ + CO32-
L'anione carbonato tende a riassociarsi con gli ioni H+ provenienti dall'acqua producendo il seguente equilibrio di idrolisi
CO32- + H2O  HCO3- + OH-
HCO3- + OH-
la costante dell'equilibrio di idrolisi è

A sua volta l'anione bicarbonato HCO3- tende a riassociarsi con gli ioni H+ provenienti dall'acqua producendo un secondo equilibrio di idrolisi basica
HCO3- + H2O  H2CO3 + OH-
H2CO3 + OH-
la cui costante di idrolisi vale

La prima costante di idrolisi ha un valore di circa quattro ordini di grandezza superiore alla seconda (kh1 >> kh2). Possiamo dunque trascurare il secondo equilibrio di idrolisi e trattare il primo come un equilibrio di dissociazione di una base debole al quale applicheremo la formula risolutiva semplificata più opportuna.
- Soluzione 3 10-2 M
Poiché il rapporto C / kh1 = 3 10-2 / 1,79 10-4 = 1.7 102 > 102 ed il prodotto C ∙ kh1 3 10-2 ∙ 1,79 10-4 = 5.4 10-6 > 10-12 possiamo usare la formula semplificata  .
.

pOH = –log(2,5 10-3) = 2,6
pH = 14 – pOH = 14 – 2,6 = 11,4
- Soluzione 10-4 M
Poiché in questo caso il rapporto C / kh1 = 10-4 / 1,79 10-4 = 0,56 < 102 non possiamo usare la formula semplificata  . Essendo tuttavia C = 10-4 > 10-7 usiamo la relazione
. Essendo tuttavia C = 10-4 > 10-7 usiamo la relazione

pOH = –log(7,15 10-5) = 4,1
pH = 14 – pOH = 14 – 4,1 = 9,9
Idrolisi acida
a) sale derivato da acido forte e base debole
Si produce un'idrolisi acida quando viene sciolto in acqua un sale il cui anione deriva da un acido forte, mentre il catione deriva da una base debole.
Prendiamo ad esempio il cloruro di ammonio NH4Cl che deriva dall'acido cloridrico HCl (acido forte con ka elevata) e dall'idrossido di ammonio NH4OH (base debole con kb bassa).
In soluzione acquosa il cloruro di ammonio è, come la maggior parte dei sali, completamente dissociato in ioni NH4+ e ioni Cl-
NH4Cl → NH4+ + Cl-
E' necessario ora analizzare in che modo tali ioni interferiscano con l'equilibrio di dissociazione dell'acqua.
Mentre lo ione cloruro Cl- , derivando da un acido forte, non ha alcuna tendenza a riassociarsi con gli ioni H+, lo ione ammonio NH4+ tende a rubare ioni OH- per ridare la base debole NH4OH.
Come al solito, per determinare le concentrazioni di equilibrio delle specie chimiche sarebbe necessario tener conto simultaneamente dei due equilibri che interferiscono: quello di dissociazione dell'acqua e quello di dissociazione della base debole
H2O  H+ + OH-
H+ + OH-
NH4OH  NH4+ + OH-
NH4+ + OH-
Le due equazioni formano un sistema che richiede la soluzione di un'equazione di grado superiore al secondo.
Soluzione esatta Nei 2 equilibri compaiono le seguenti 4 incognite. 1) [H+] 2) [OH–] 3) [NH4OH] 4) [NH4+]
Dobbiamo pertanto scrivere 4 equazioni indipendenti nelle 4 incognite.
la terza si ottiene dal bilancio delle cariche (la somma di tutte le cariche positive deve essere uguale alla somma di tutte le cariche negative)
la quale, ricordando che il sale è completamente dissociato e quindi [Cl-] = C, diventa
la quarta si ricava infine dal bilancio di massa. Ricordando che il sale è completamente dissociato in C mol/L di ipoclorito Cl– e C mol/L di ioni ammonio NH4+, dopo l’idrolisi la somma dell’idrossido di ammonio indissociato che si forma e dello ione ammonio residuo all’equilibrio dovrà essere pari alla concentrazione iniziale C e potremo quindi scrivere
utilizzando le 4 equazioni ed effettuando le opportune sostituzioni si ottiene
un’equazione di terzo grado che ci permette di calcolare il valore esatto della concentrazione totale di ioni H+ per una soluzione di un sale formato da un acido forte ed una base debole. |






Se la concentrazione iniziale del sale è sufficientemente elevata è comunque possibile, come per l'idrolisi basica, ricorrere ad una descrizione semplificata del fenomeno, scrivendo il seguente equilibrio di idrolisi
NH4+ + H2O  NH4OH + H+
NH4OH + H+
la cui costante di equilibrio vale naturalmente

Tale relazione mette in evidenza come l'equilibrio di idrolisi acida è tanto più spostato verso destra quanto più la base è debole (maggiore tendenza ad associarsi liberando ioni H+). Di conseguenza la soluzione risulterà essere tanto più acida quanto più piccolo è il valore della kb (e più elevato il valore della kh).
Esempio
Calcoliamo il pH di una soluzione 0,5 M di cloruro di ammonio, sapendo che la kb dell'ammoniaca è pari 1,8.10-5.
Il cloruro di ammonio si dissocia completamente in 0,5 mol/L di ioni NH4+ e 0,5 mol/L di ioni Cl-.
NH4Cl → NH4+ + Cl-
Come abbiamo visto, lo ione ammonio tende a riassociarsi con gli ioni OH- provenienti dall'acqua producendo il seguente equilibrio di idrolisi
NH4+ + H2O  NH4OH + H+
NH4OH + H+
Calcoliamo la costante dell'equilibrio di idrolisi

Indichiamo con x la quantità di NH4+ che si riassocia per dare x mol/L di NH4OH, mentre vengono contemporaneamente liberate x mol/Ldi ioni H+,
|
iniziale |
d'equilibrio |
[NH4+] |
0,5 |
0,5 - x |
[NH4OH] |
0 |
x |
[H+] |
0 |
x |
Esprimiamo le concentrazioni di equilibrio in funzione della costante di idrolisi

risolvendo l'equazione otteniamo
x = [H+]= [NH4OH] = 1,67.10-5 mol/L pH = 4,8
(l’equazione di terzo grado fornisce [H+] = 1,67.10-5 mol/L)
Si noti come anche in questo caso era possibile ricorrere ad una soluzione semplificata in quanto la concentrazione iniziale del sale è sufficientemente elevata e la kh è sufficientemente piccola da far ritenere che la quantità di ioni OH- che si riassocia sia trascurabile rispetto alla concentrazione iniziale dello ione ammonio.
Il prodotto kh∙C = 5.56 10-10 ∙ 0,3 = 1.7 10-10 ed essendo maggiore di 10-12 ci permette di usare la prima formula semplificata per il calcolo del pH
Se dunque trascuriamo la x nella differenza a denominatore, possiamo usare la seguente relazione semplificata

dove Csale è la concentrazione iniziale del sale
Idrolisi acida di un catione metallico
I cationi dei metalli (in modo particolare i metalli di transizione) formano in soluzione degli ioni idratati (composti di coordinazione) che si comportano come acidi. Le soluzioni di tali sali sono tutte più o meno acide ed il processo che avviene è analogo a quello descritto per l'idrolisi acida. In tal caso però il valore della costante di idrolisi viene espresso come ka dello ione idratato (acquoione).
Ad esempio le soluzioni di cloruro ferrico FeCl3 sono molto acide. Potremmo descrivere il fenomeno dicendo che mentre lo ione cloruro Cl- non si riassocia per dare l'acido cloridrico forte, lo ione ferrico Fe3+ da luogo al seguente equilibrio di idrolisi
Fe3+ + H2O  FeOH2+ + H+
FeOH2+ + H+
Tale reazione viene però comunemente indicata come dissociazione acida dello ione ferrico e la costante di reazione è nota come ka e non come kh.

In realtà la reazione reale non coinvolge lo ione ferrico, ma lo ione idratato (ione esaacquoferro(III)) e questo vale per la maggior parte dei cationi metallici con proprietà acide.
Fe(H2O)63+  [Fe(OH)(H2O)5]2+ + H+
[Fe(OH)(H2O)5]2+ + H+
Lo ione complesso esaacquoferrico si comporta come un acido dissociandosi in uno ione H+ e in uno ione complesso pentaacquo-idrosso-ferrico, responsabile quest'ultimo della comune colorazione verde-giallastro delle soluzioni dei sali ferrici (lo ione esaacquoferrico ha una colorazione porpora chiaro).
I due equilibri precedenti sono comunque equivalenti.
Esempio
Calcoliamo la concentrazione di ioni H+ in una soluzione 10-3 M di FeCl3 (ka = 6,3 10-3) ed in una soluzione 10-7 M del medesimo sale.
Fe3+ + H2O  FeOH2+ + H+
FeOH2+ + H+
Trattiamo l’equilibrio di idrolisi acida come un normale equilibrio di dissociazione di un acido debole ed individuiamo la formula semplificata da applicare alla soluzione 10-3 M.
Poiché C/ka = 10-3 / 6.3 10-3 = 0.16 < 102 e C = 10-3 > 10-7 utilizziamo la relazione

Individuiamo ora la formula semplificata da applicare alla soluzione 10-7 M. Poiché C = 10-7 ≤ 10-7 e ka = 6,3 10-3 >10-7 utilizziamo la relazione

Le molecole d’acqua coordinate ai cationi metallici hanno proprietà acide più spiccate delle molecole d’acqua libere di muoversi nella soluzione. Il fenomeno è da porre in relazione con l’indebolimento del legame O-H delle molecole coordinate a seguito dello spostamento della nube elettronica verso l’ossigeno, a sua volta causato dalla presenza dell’interazione tra i doppietti elettronici dell’ossigeno e la carica positiva dello ione metallico.
Più forte è l’interazione e più è acido il catione metallico.
L’intensità dell’interazione dipende principalmente, dalla carica del catione (Z) dal raggio ionico (r) e dall’elettronegatività dell’elemento considerato.
Si osserva che all’aumentare della carica e al diminuire del raggio ionico l’acidità aumenta.
Per cationi con elettronegatività (secondo Pauling) inferiore a 1,5 si trova una proporzionalità diretta tra il valore del pka ed il rapporto Z2/r. Se il raggio ionico viene espresso in picometri si ha
pka = 15.14 - 88.16(Z2/r)
Inoltre, a parità di Z2/r, i cationi con elettronegatività superiore a 1,5 sono in genere più acidi. Per essi vale la relazione
pKa = 15.14 - 88.16[(Z2/r) + 0. 096(cp-1.50)]
con cp = Elettronegatività secondo Pauling
In altre parole la coordinazione modifica le proprietà acido-base dell’acqua.
Il legante H2O (pka = 14.0) coordinato agli ioni Ca2+ o Zn2+ modifica il suo valore di pka a 12.8 e 9.5, rispettivamente:
[Zn(H2O)6]2+ + H2O ⇄ [Zn(H2O)5(OH)]+ + H3O+ ka = 3.3 10-10
Questo effetto è del tutto generale e, oltre all’acqua quale legante, vale per molti altri leganti (tioli, fenoli, alcoli, acido fosforico e acidi carbossilici).
La carica positiva del catione metallico stabilizza infatti la base coniugata di acidi protici attraverso la coordinazione al centro metallico.
Ad esempio l’acido acetico in acqua ha pKa = 4.7, ma in presenza dello ione Ni2+ (0.1 M) il pka diventa 4.0 e se lo ione è Cu2+ il pka diventa 3.0.
La coordinazione dello ione acetato al centro metallico sposta l’equilibrio di dissociazione dell’acido a destra e l’acido acetico diventa un acido più forte.
I cationi metallici possono essere classificati in gruppi a seconda del valore di Z2/r.
Relazione tra il rapporto Z2/r e l’ acidità dei cationi metallici |
||||
Z2/r |
χp |
Acidità |
Intervallo di pKa |
Esempi |
< 0.01 |
<1.5 |
Non acidi |
14.0 ÷ 15.0 |
Maggior parte dei cationi alcalini |
< 0.01 |
>1.5 |
Acidi debolissimi |
11.5 ÷ 14.0 |
Tl+ |
0.01 ÷ 0.04 |
<1.5 |
Maggior parte dei cationi alcalino-terrosi |
||
0.01 ÷ 0.04 |
>1.5 |
Acidi deboli |
6 ÷ 11.5 |
Maggior parte dei cationi 2+ del blocco “d” |
0.04 ÷ 0.10 |
<1.5 |
Tutti i cationi 3+ del blocco “f” |
||
0.01 ÷ 0.10 |
>1.5 |
Acidi moderati |
1 ÷ 6 |
Maggior parte dei cationi 3+ del blocco “d” |
0.10 ÷ 0.16 |
<1.5 |
Maggior parte dei cationi 4+ del blocco “f” |
||
0.10 ÷ 0.16 |
>1.5 |
Acidi forti |
– 4 ÷ 1 |
Maggior parte dei cationi 4+ del blocco”d” |
0.16 ÷ 0.22 |
<1.5 |
|
||
> 0.16 |
>1.5 |
Acidi fortissimi |
< – 4 |
|
> 0.22 |
<1.5 |
|
||
In relazione alla diversa acidità del catione metallico, l’idrolisi può proseguire oltre la formazione dell’idrosso-catione, attraverso una serie di tappe caratteristiche che portano alla formazione di un idrossido (in genere insolubile) e, se il metallo è molto acido, di un idrosso-anione ed infine un osso-anione.
Ad esempio per l’Alluminio si può avere la seguente progressione
Al3+ + 6 H2O ⇄ Al(H2O)63+
Catione metallico catione idratato (acquo-ione)
1) Al(H2O)33+ + H2O ⇄ Al(H2O)5(OH)2+ + H3O+ ka = 1,4 10-5
Acquo-ione Idrosso-catione
2) Al(H2O)5(OH)2+ ⇄ Al(H2O)(OH)3 + 2 H3O+
Idrosso-catione Idrossido (idratato)
3) Al(H2O)(OH)3 + H2O ⇄ Al(OH)4– + H3O+
Idrossido Idrosso-anione
4) Al(OH)4– + 4 H2O ⇄ AlO45– + 4 H3O+
Idrosso-anione Ossoanione
In generale, la forma con cui un catione metallico è presente in soluzione dipende dunque dalla sua acidità, ma è comunque possibile spostare gli equilibri tramite variazioni di pH.
Un catione moderatamente acido come l’alluminio (pka = 4,85) non è in grado di completare la sequenza, ma è ovviamente possibile spostare gli equilibri verso destra sia aggiungendo ioni OH- (aumentando il pH) che rimuovono gli ioni H3O+ (formando acqua) o verso sinistra aggiungendo ioni H+ (diminuendo il pH).
I cationi, con un rapporto carica-raggio molto grande Z2/r >> 0.22, non esistono in soluzione acquosa come tali, nemmeno a pH fortemente acido. Sono presenti invece i loro ossoanioni.
Il catione Al3+ inizia a precipitare come idrossido quando il pH > 5, raggiunge un minimo di solubilità per pH = 7 e si ridiscioglie come idrosso-anione e osso-anione per pH > 12.

Si tratta di un comportamento caratteristico degli idrossidi anfoteri insolubili, i quali ridiventano solubili sia quando si abbassa il pH, comportandosi come basi, che quando si alza il pH, comportandosi come acidi.
Al(OH)3(s) + 3 H3O+ + 3 H2O ⇄ Al(H2O)33+ (comportamento basico)
Idrossido insolubile catione-idratato solubile
Al(OH)3(s) + OH– ⇄ Al(OH)4– (comportamento acido)
Idrossido insolubile Idrosso-anione solubile
L’idrolisi come reazione acido-base di Brønsted
Come avremo modo di vedere successivamente, le reazioni di idrolisi possono essere descritte come reazioni acido-base secondo la teoria di Brønsted-Lowry.
- Un acido di Brønsted è una sostanza in grado di cedere ioni H+ (protoni)
- Una base di Brønsted è una sostanza in grado di acquistare ioni H+ (protoni)
Secondo la teoria di Brønsted e Lowry una reazione acido-base consiste dunque nel trasferimento di un protone da un acido ad una base.

H-A + :B → :A- + H-B+
acido base
Ogni acido di Brønsted, cedendo uno ione H+, genera una specie chimica che può rilegarsi ad esso. La specie generata è detta base coniugata dell'acido.
Ogni base di Brønsted, acquistando uno ione H+, genera una specie chimica che può rilasciarloLa specie generata è detta acido coniugato della base.

Nella reazione precedente sono presenti le due coppie acido/base
HA/A- e HB+/B
L'acqua viene vista come un composto anfotero, potendo sia cedere che acquistare ioni H+,
H2O + H2O  OH¯ + H3O+
OH¯ + H3O+
acido base base acido
coniugata coniugato
presenta due coppie coniugate acido/base
H3O+ / H2O H2O / OH¯
acido base acido base
Ovviamente se un acido è forte, con una elevata tendenza a cedere ioni H+, la sua base coniugata sarà debole, manifestando una scarsa tendenza ad acquistare ioni H+ (e viceversa).
La forza di un acido o di una base viene misurata in relazione alla forza dell’acqua, cioè alla tendenza che l’acqua manifesta a cedere ed acquistare ioni H+.
La forza di un acido HA, misura la sua tendenza a cedere ioni H+ all’acqua secondo l’equilibrio
HA + H2O  A¯ + H3O+
A¯ + H3O+
acido base
La costante di equilibrio sarà

Ed inglobando la concentrazione dell’acqua (considerata costante e pari a 55,55 mol/L) nella costante di equilibrio otteniamo la ka.

Definiamo ora la basicità della sua base coniugata (A-), misurando la sua tendenza ad acquistare ioni H+ dall’acqua.
A¯ + H2O  HA + OH¯
HA + OH¯
base acido
Si noti come l’equilibrio acido-base della base coniugata A¯ non sia altro che l’equilibrio che abbiamo descritto in precedenza come una idrolisi basica
La costante di equilibrio sarà

Ed analogamente a quanto abbiamo fatto in precedenza, definiamo anche per esso la kb, inglobando la concentrazione dell’acqua nella costante di equilibrio

Si noti come la kb della base coniugata coincida con la costante di idrolisi kh
Moltiplicando ora numeratore e denominatore per la concentrazione degli ioni idronio, otteniamo, la relazione esistente tra la kappa acida di un acido e la kappa basica della sua base coniugata.

Dunque per un acido e la sua base coniugata vale la relazione
kw = 10-14 = ka · kb.
Le rispettive costanti di dissociazione acida e basica sono dunque inversamente proporzionali. Più forte è un acido e più debole è la sua base coniugata.
Ovviamente la medesima dimostrazione può essere fatta per una base ed il suo acido coniugato.
Idrolisi neutra
(sale derivato da un acido forte e da una base forte)
Si produce un'idrolisi neutra quando viene sciolto in acqua un sale il cui anione deriva da un acido forte ed il cui catione deriva da una base forte.
Prendiamo ad esempio il nitrato di potassio KNO3 che deriva dall'acido nitrico HNO3 (acido forte con ka elevata) e dall'idrossido di potassio KOH (base forte con kb elevata).
In soluzione acquosa il nitrato di potassio è, come la maggior parte dei sali, completamente dissociato in ioni K+ e ioni NO3-
KNO3 → K+ + NO3-
Ora, poiché sia lo ione K+ che lo ione NO3- non hanno alcuna tendenza a riassociarsi per ridare la base e l'acido, entrambi forti, da cui derivano, l'equilibrio di dissociazione dell'acqua non viene disturbato e la soluzione rimane neutra.
Idrolisi di un sale derivante da un acido debole e da una base debole
In tal caso, dopo che il sale si è dissociato in acqua, sia il catione che l'anione prodotti tendono a riformare, naturalmente in misura diversa, la base debole e l'acido debole da cui provengono. Il pH della soluzione sarà naturalmente acido se kb < ka, mentre sarà basico in caso contrario.
Prendiamo ad esempio il cianuro di ammonio NH4CN, che deriva dall'idrossido di ammonio NH4OH (base debole, kb = 1,8.10-5) e dall'acido cianidrico HCN (acido debole, ka = 4,9.10-10).
Per calcolare il pH di tale soluzione sarebbe necessario tener conto simultaneamente dei seguenti tre equilibri
1) H2O  H+ + OH-
H+ + OH-
2) NH4OH  NH4+ + OH-
NH4+ + OH-
3) HCN  H+ + CN-
H+ + CN-
Poiché la costante di dissociazione dell'acido cianidrico è inferiore di quella dell'ammoniaca acquosa (ka < kb) possiamo prevedere che l'acido cianidrico si riassoci in percentuale maggiore rispetto all'idrossido di ammonio (il 3° equilibrio risulta cioè più spostato verso sinistra rispetto al 2°). La soluzione sarà pertanto basica.
La soluzione esatta del problema richiederebbe naturalmente la risoluzione di un sistema di equazioni in cui compaiano tutte le condizioni relative ai tre equilibri.
Soluzione esatta Nei tre equilibri compaiono le seguenti 6 incognite. 1) [H+] 2) [OH–] 3) [HCN] 4) [CN–] 5) [NH4OH] 6) [NH4+] Dobbiamo pertanto scrivere 6 equazioni indipendenti nelle 6 incognite.
la quarta si ottiene dal bilancio delle cariche (la somma di tutte le cariche positive deve essere uguale alla somma di tutte le cariche negative)
la quinta e la sesta si ricavano infine dal bilancio di massa per l’acido e per la base.
utilizzando le 6 equazioni ed effettuando le opportune sostituzioni si ottiene un’equazione di quarto grado che ci permette di calcolare il valore esatto della concentrazione totale di ioni H+ per una soluzione di un sale formato da un acido debole ed una base debole. |







La risoluzione approssimata prende invece in considerazione il seguente equilibrio di idrolisi
NH4+ + CN– + H2O  NH4OH + HCN
NH4OH + HCN
C – x C – x x x
secondo il quale per ogni ione OH– che si riassocia con uno ione NH4+, uno ione H+ si riassocia con uno ione CN–.
Si può facilmente dimostrare che la costante di tale equilibrio vale

Tenendo presente che le concentrazioni iniziali di NH4+ e di CN– sono pari alla concentrazione C del sale completamente dissociato, sarà quindi possibile calcolare le concentrazioni di equilibrio delle quattro specie chimiche.

Come si può osservare si ottengono delle concentrazioni uguali per le specie dissociate (x = [NH4+] = [CN–]) e per le specie indissociate ( C – x = [NH4OH] = [HCN]) e quindi risulteranno uguali anche i loro rapporti

se dunque dalla relazione di equilibrio della base esplicitiamo il rapporto tra le specie dissociata e quella indissociata

e lo sostituiamo nella relazione di equilibrio dell’acido, otteniamo

ricordando infine che [OH–] = kw/[H+], sostituendo otteniamo

e quindi
 mol/L
mol/L
che corrisponde ad un pH = 9.28 (basico).
Si noti come la concentrazione degli ioni [H+], e quindi anche il pH, risulti indipendente (almeno nell’approssimazione che stiamo considerando) dalla concentrazione C del sale.
Si noti anche che il rapporto kw/kb è pari alla ka dell’acido coniugato (NH4+) della base (NH4OH) e quindi la formula semplificata per il calcolo del pH di una soluzione contenente un sale formato da un acido debole ed una base debole può anche essere scritta

con
ka = kappa acida dell’acido debole
kac = kappa acida dell’acido coniugato della base debole
Si tenga infine presente che se il sale è molto diluito (C ≤ 10-4 M) la formula semplificata non fornisce un risultato soddisfacente e la concentrazione degli ioni H+ risente della concentrazione del sale. In tal caso è conveniente calcolare il pH trascurando l’idrolisi dello ione che presenta la costante di idrolisi più piccola ed utilizzando per l’idrolisi dell’altro ione le formule trovate per il calcolo del pH di acidi (o basi) deboli monoprotici (a seconda del valore di C e kh).
Esempio
Calcoliamo la concentrazione di ioni H+ di una soluzione 10-2 M di ipoclorito di ammonio NH4ClO (HClO ka = 3 10-8 – NH3 kb = 1,8 10-5) e di una soluzione 10-6 M del medesimo sale.
Sono presenti i seguenti equilibri di idrolisi
1) idrolisi acida NH4+ + H2O 
2) idrolisi basica ClO- + H2O 
Essendo la costante di idrolisi basica più elevata della costante di idrolisia acida ( ), la soluzione risulterà basica. Se applichiamo la formula approssimata otteniamo il medesimo valore di [H+] sia per la soluzione 10-2 M che per la soluzione 10-6 M.
), la soluzione risulterà basica. Se applichiamo la formula approssimata otteniamo il medesimo valore di [H+] sia per la soluzione 10-2 M che per la soluzione 10-6 M.

Se applichiamo l’equazione di quarto grado che ci fornisce il risultato esatto troviamo:
- per la soluzione 10-2 M una concentrazione idrogenionica [H+] = 4,09 10-9 mol/L
- per la soluzione 10-6 M una concentrazione idrogenionica [H+] = 2,30 10-8 mol/L
Come si può osservare la soluzione 10-6 M presenta una concentrazione degli ioni H+ inferiore di circa un ordine di grandezza rispetto alla soluzione 10-2 M.
Trascuriamo allora l’equilibrio di idrolisi acida, la cui costante di equilibrio è di circa 3 ordini di grandezza inferiore rispetto all’equilibrio di idrolisi basica.
Applichiamo ora all’equilibrio di idrolisi basica una delle quattro formule risolutive semplificate per il calcolo del pH di acidi e basi deboli monoprotici.
Verifichiamo quali condizioni di applicabilità sono soddisfatte
1) Poiché C·kh(B) = 10-6 x 3,3 10-7 = 3,3 10-13 < 10-12 non possiamo utilizzare la relazione

2) Poiché kh(B) = 3,3 10-7 > 10-7 non possiamo utilizzare la relazione

3) Poiché C = 10-6 > 10-7 non possiamo utilizzare la relazione

4) Poiché C = 10-6 > 10-7 e C/k = 10-6 / 3.3 10-7 = 3 < 102, possiamo utilizzare la relazione

e dunque

Idrolisi di elettroliti anfoteri (anfoliti)
Un elettrolita anfotero o anfolita è un elettrolita che può comportarsi sia come acido (cedendo protoni) che come base (acquistando protoni). Il suo comportamento risulta quindi analogo a quello di un sale formato da un acido debole ed una base debole (vedi paragrafo precedente).
Consideriamo ad esempio una soluzione di Idrogeno Carbonato di Sodio NaHCO3 di concentrazione C = 10-2 M. In soluzione è completamente dissociato in C ioni Na+ e C ioni HCO3–. L’acido carbonico H2CO3, essendo un acido debole biprotico, presenta due costanti di dissociazione pari a ka1 = 4.3 10-7 e ka2 = 5.6 10-11.
NaHCO3 → Na+ + HCO3–
-C +C +C
L’anione bicarbonato HCO3– in soluzione si comporta sia come un acido, dando l’equilibrio di seconda dissociazione, con ka2 = 5.6 10-11
HCO3–  H+ + CO32-
H+ + CO32-
sia come una base, dando idrolisi basica, con kh = kw/ ka1 = 10-14/4.3 10-7= 2.36 10-8 (come abbiamo visto in precedenza la reazione può anche essere considerata una reazione acido-base con HCO3– che rappresenta la base coniugata (kbc = kw/ ka1 = 10-14/4.3 10-7= 2.36 10-8) dell’acido carbonico)
HCO3– + H2O  H2CO3 + OH-
H2CO3 + OH-
Per calcolare il pH di tale soluzione sarebbe necessario tener conto simultaneamente dei precedenti due equilibri e dell’equilibrio di dissociazione dell’acqua.
Poiché la costante di seconda dissociazione dell'acido carbonico (ka2 = 5.6 10-11) è inferiore alla sua costante di idrolisi (kh = kw/ ka1 = 2.36 10-8) possiamo prevedere che l'anione bicarbonato si riassoci in percentuale maggiore (idrolisi basica) rispetto a quanto si dissoci (dissociazione acida). La soluzione sarà pertanto basica.
La soluzione esatta del problema richiederebbe naturalmente la risoluzione di un sistema di equazioni in cui compaiano tutte le condizioni relative ai tre equilibri.
Soluzione esatta Nei tre equilibri compaiono le seguenti 5 incognite. 1) [H+] 2) [OH–] 3) [H2CO3] 4) [HCO3–] 5) [CO32–] Dobbiamo pertanto scrivere 5 equazioni indipendenti nelle 5 incognite. 1) 2) 3) la quarta si ottiene dal bilancio delle cariche (la somma di tutte le cariche positive deve essere uguale alla somma di tutte le cariche negative) e, ricordando che il sale è completamente dissociato e quindi che [Na+] = C la quinta si ricava infine dal bilancio di massa. 5) utilizzando le 5 equazioni ed effettuando le opportune sostituzioni in funzione della concentrazione di ioni H+ si ottiene
con x = [H+] un’equazione di quarto grado che ci permette di calcolare il valore esatto della concentrazione totale di ioni H+ per una soluzione di un anfolita. |







Per un calcolo approssimato sottraiamo membro a membro il bilancio di carica ed il bilancio di massa, ottenendo
6) 
dalla 1) esplicitiamo la concentrazione dell’acido indissociato H2CO3 e sostituiamola nella 6)
7) 
dalla 2) ricaviamo la concentrazione dell’anione carbonato CO32- e sostituiamola nella 7)
8) 
dal prodotto ionico dell’acqua ricaviamo la concentrazione degli ioni OH- e sostituiamo nella 8)
9) 
Se ora assumiamo che, sia l’equilibrio di idrolisi che quello di dissociazione acida, avendo in genere costanti piccole, siano molto spostati verso sinistra, la concentrazione di HCO3- sarà prevalente rispetto a quelle di CO32- e H2CO3
[HCO3-] >> [CO32-] + [H2CO3]
e quindi
[HCO3-] + [CO32-] + [H2CO3] ≈ [HCO3-]
ciò significa che la concentrazione di equilibrio di HCO3- è praticamente uguale alla sua concentrazione iniziale. Dal bilancio di massa si avrà infatti

che, sostituita nella 9), ci dà


10) 
Sostituendo i valori otteniamo

La 10) può essere ulteriormente semplificata, dividendo numeratore e denominatore per C otteniamo
11) 
se ora, come spesso avviene, sono verificate le seguenti condizioni
C >> ka1 e 
e quindi

la 11) diventa
12) 
Eseguendo il logaritmo decimale negativo di entrambi i membri si ha
pH = ½ (pka1 + pka2)
La 12) equivale a prendere in considerazione il seguente equilibrio, in cui si assume che l’equilibrio di seconda dissociazione dell’acido sia sincronizzato con l’equilibrio di idrolisi
HCO3– + HCO3–  H2CO3 + CO32-
H2CO3 + CO32-
C – x C – x x x
In altre parole, per ogni ione HCO3– che si dissocia in CO32- e H+, un altro ione HCO3– si riassocia con uno ione H+ per dare . H2CO3
Come si può osservare, nell’approssimazione considerata si assume che all’equilibrio sia
[H2CO3] = [CO32–]
ora esplicitiamo la concentrazione di HCO3- dalla relazione di equilibrio della prima dissociazione acida

e lo sostituiamo nella relazione di equilibrio della seconda dissociazione acida

ricordando infine che [H2CO3] = [CO32–], otteniamo

e quindi
 mol/L
mol/L
che corrisponde ad un pH = 8.31 (basico).
Si noti come, anche in questo caso come nel paragrafo precedente, la concentrazione degli ioni H+, e quindi anche il pH, risulti indipendente (almeno nell’approssimazione che stiamo considerando) dalla concentrazione C del sale.
Si noti infine che la formula semplificata per il calcolo del pH di una soluzione contenente un elettrolita anfotero è analoga a quella trovata nel paragrafo precedente per un sale formato da un acido debole ed una base debole.

con
ka1 = costante di prima dissociazione acida
ka2 = costante di seconda dissociazione acida
kh = costante di idrolisi
Se il sale deriva da un acido debole triprotico H3A (ad esempio l’acido fosforico H3PO4), le formule per il calcolo del pH saranno
- Per un sale del tipo MH2A D M+ + H2A-

- Per un sale del tipo M2HA D 2M+ + HA2-

Soluzioni tampone
Si dicono tamponate quelle soluzioni acquose che conservano praticamente immutato il proprio pH anche dopo l'aggiunta di piccole quantità di acidi o basi.
I tamponi hanno una straordinaria importanza biologica, infatti moltissime soluzioni biologiche (sangue, linfa, succhi gastrici etc) richiedono il mantenimento di un pH costante.
L'effetto tampone si ottiene sciogliendo in acqua un acido debole con un suo sale o una base debole con il relativo sale.
Consideriamo ad esempio una soluzione contenente CA mol/L di un generico acido debole HA e CS mol/L di un suo sale di sodio NaA.
L'acido debole è presente in soluzione quasi totalmente indissociato essendo il suo equilibrio fortemente spostato verso sinistra.
Il sale dell'acido, come la maggior parte dei sali, è invece completamente dissociato

Essendo un elettrolita forte il sale rimane completamente dissociato anche in presenza dell'acido debole.
L'equilibrio di dissociazione dell'acido viene invece disturbato dalla presenza degli ioni A- prodotti dalla dissociazione del sale, ed il suo equilibrio retrocede spostandosi ulteriormente verso sinistra.
Per calcolare il pH di una soluzione tampone determiniamo le concentrazioni di equilibrio. Poiché il sale si dissocia completamente esso produce una quantità di ioni A- pari alla sua concentrazione iniziale.Possiamo allora scrivere

Ora descriviamo l’equilibrio di dissociazione dell’acido debole HA che si dissocia, in presenza di [A-] = Csale, in x ioni H+ e x ioni A-
|
iniziale |
d'equilibrio |
[HA] |
CA |
CA - x |
[H+] |
0 |
x |
[A-] |
CS |
CS + x |
Esprimiamo ora le concentrazioni di equilibrio in funzione della ka

Essendo l'acido molto debole la concentrazione di equilibrio degli ioni H+ è molto piccola ed in genere trascurabile rispetto alle concentrazioni iniziali dell'acido (CA) e del sale (CS), specialmente se queste sono sufficientemente elevate.
Per questo motivo è possibile trascurare la x nella somma a numeratore e nella differenza a denominatore. La relazione diventa quindi

dalla quale otteniamo

Calcolando il logaritmo negativo di entrambi i membri otteniamo finalmente la relazione per il calcolo del pH in soluzioni tampone (equazione di Henderson-Hasselbach)

Se ad esempio costruiamo una soluzione tampone in cui sono presenti 10-2 mol/l di acido fluoridrico (ka = 3,53.10-4) e 10-1 mol/l di fluoruro di sodio, la soluzione risulta tamponata a

Naturalmente, se la soluzione tampone è costruita mescolando una base debole con un suo sale, si avrà


E’ tuttavia possibile e conveniente utilizzare un’unica formula risolutiva per il calcolo del pH di un sistema tampone

Se ad esempio costruiamo un tampone costituito da una soluzione NH3 10-1 M / NH4Cl 10-2 M, lo ione ammonio NH4+ rappresenta l’acido coniugato dell’ammoniaca kb = 1,8 10-5 (ed ovviamente l’ammoniaca è la base coniugata dell’acido NH4+)
NH3 + H2O D NH4+ + OH-
La relazione per il calcolo del pH diventa

La ka è ovviamente quella dell’acido NH4+ e si calcola come ka = kw/kb = 10-14/ 1,8 10-5 = 5,6 10-10.
pka = - log (5,6 10-10) = 9,26 e dunque

Meccanismo d'azione di una soluzione tampone
Cerchiamo ora di capire per qual motivo una soluzione tampone è in grado di mantenere praticamente invariato il suo pH.
Come si può osservare in una soluzione tampone sono presenti in elevata concentrazione sia l'acido debole indissociato che il suo anione (quest'ultimo prodotto dalla dissociazione del sale). Si noti che se fosse presente solo l'acido debole la concentrazione del suo anione sarebbe invece molto bassa in quanto l'acido è poco dissociato.
Ora se aggiungiamo alla soluzione un acido forte, gli ioni H+ prodotti dalla sua completa dissociazione tendono a combinarsi con gli ioni A- della soluzione tampone per dare l'acido debole indissociato HA. Naturalmente il processo risulta efficiente solo se in soluzione è presente una elevata concentrazione di ioni A-.
Se invece viene addizionata una base forte gli ioni OH- prodotti dalla sua completa dissociazione si uniscono agli ioni H+ prodotti dall'acido debole per dare acqua indissociata (l'equilibrio di dissociazione dell'acqua è infatti molto spostato verso sinistra, kw piccola). Naturalmente man mano che gli ioni H+ vengono sottratti all'equilibrio dell'acido debole, quest'ultimo, per il principio di Le Chatelier, si dissocia ulteriormente fornendo altri ioni H+ che vanno a neutralizzare gli ioni OH-.
Anche in questo caso il processo risulta efficiente solo se nella soluzione tampone è presente una elevata quantità di acido debole indissociato in grado di fornire ioni H+.
Il meccanismo d'azione di una soluzione tampone si fonda dunque sulla presenza contemporanea di una elevata concentrazione di acido debole e del suo anione.
Osservando la relazione per il calcolo del pH di una soluzione tampone si può osservare come il pH dipenda dal valore della ka e dal rapporto tra la concentrazione dell'acido e del suo sale.
Una soluzione tampone può essere più o meno efficiente. L'efficienza di una soluzione tampone può essere definita sulla base della diversa capacità di mantenere più o meno inalterato il pH a fronte di un'aggiunta di una medesima quantità di acido o base forte. Sono naturalmente più efficienti le soluzioni tampone che riescono a far variare il pH in misura minore.
Si può dimostrare che l'efficienza di una soluzione tampone cresce al crescere della sua concentrazione complessiva.
A parità di concentrazione totale è inoltre più efficiente la soluzione tampone che presenta un rapporto tra la concentrazione dell'acido e quella del sale più vicino all'unità. L'efficienza massima si ottiene quando Cacido/Csale = 1. Come si può facilmente verificare in tal caso la soluzione risulta tamponata ad un pH = pk.
Ad esempio possiamo affermare che la soluzione tampone presentata nell'esempio precedente, in cui era stato utilizzato acido fluoridrico (ka = 3,53.10-4), raggiunge la sua massima efficienza quando viene fatta lavorare a pH = pk = -log 3,53.10-4 = 3,45.
Verifichiamo ora con un esempio le condizioni di efficienza delle soluzioni tampone.
a) Variazione della concentrazione totale in una soluzione tampone
Prendiamo in considerazione 2 soluzioni tamponate, la prima più diluita in cui Csale = Cacido = 0,4 mol/L, la seconda più concentrata, con Csale = Cacido = 0,8 mol/L. Entrambe le soluzioni sono tamponate ad un pH = pk in quanto Cacido/Csale = 1.
Osserviamo ora come l'aggiunta di 0,2 mol/l di un acido forte ad entrambe le soluzioni faccia variare il pH della soluzione più diluita in modo più evidente.
L'acido forte è completamente dissociato e libera pertanto 0,2 mol/L di ioni H+, i quali si riassociano con altrettante moli di anione per ridare l'acido debole. All'equilibrio la concentrazione dell'anione (Csale) sarà quindi diminuita di 0,2 mol/L, mentre la concentrazione dell'acido debole (Cacido), sarà aumentata in egual misura. Il nuovo pH sarà pertanto
per il 1° tampone 
per il 2° tampone 
La soluzione tampone più concentrata vede cambiare il suo pH di 0,22 unità contro le 0,48 unita della soluzione più diluita.
b) Variazione del rapporto Cacido/Csale in una soluzione tampone.
Prendiamo ora in considerazione due soluzioni tampone che presentano la stessa concentrazione totale, ma diverso rapporto Acido/Sale. Sia la ka = 10-5. La prima avrà Csale = Cacido = 0,8 mol/L, la seconda Cacido = 1,2 mol/L e Csale = 0,4 mol/L. Aggiungiamo ad entrambe le soluzioni 0,2 mol/L di un acido forte e calcoliamo le relative variazioni di pH
per il 1° tampone il pH passa
da 
a 
con una diminuzione del 4,4%
per il 2° tampone il pH varia
da 
a 
con una diminuzione del 8,2%
Il sangue umano è una soluzione tamponata a pH 7,4. Tra i diversi sistemi tampone presenti nel sangue, il più importante è quello costituito dall'acido carbonico (che si produce dalla reazione dell'anidride carbonica respiratoria con l'acqua) e dallo ione bicarbonato (H2CO3/HCO3-). In effetti lo ione bicarbonato presenta una concentrazione plasmatica circa 10 volte superiore di quella dell'acido carbonico. Il tampone plasmatico H2CO3/HCO3- è quindi lontano dalle condizioni di maggior efficienza. La maggior concentrazione di ioni bicarbonato permette però al sangue di tamponare più facilmente sostanze acide, che rappresentano i principali cataboliti versati nel sangue (acido lattico, acidi urici etc).
In altre parole l'organismo si difende meglio dagli squilibri legati ad eccesso di acidi piuttosto che ad eccesso di basi.
Sapendo che la costante di dissociazione acida dell’acido carbonico è pari a ka = 4,3 10-7, possiamo scrivere

fonte: http://www.pianetachimica.it/didattica/documenti/Chimica_Generale.doc
Chimica generale 9
Visita la nostra pagina principale
Chimica generale 9
Termini d' uso e privacy