Biochimica
Biochimica
I riassunti, le citazioni e i testi contenuti in questa pagina sono utilizzati per sole finalità illustrative didattiche e scientifiche e vengono forniti gratuitamente agli utenti.
Le informazioni di medicina e salute contenute nel sito sono di natura generale ed a scopo puramente divulgativo e per questo motivo non possono sostituire in alcun caso il consiglio di un medico (ovvero un soggetto abilitato legalmente alla professione)
APPUNTI DI BIOCHIMICA PER CORSO AGGIORNAMENTO
EDUCATORI PROFESSIONALI
Prof.ssa Elena Zocchi
Gli enzimi
Un enzima è un catalizzatore (acceleratore) di reazioni biologiche. L’enzima accelera il raggiungimento dell’equilibrio* di una reazione chimica (spontanea!) senza spostarlo. Quindi accelera (da 107 a 1014 volte!) della stessa entità sia la reazione in un senso che quella in senso opposto:
A + B « C + D
Tutti gli enzimi sono proteine (alcuni RNA hanno attività catalitica).
*L’equilibrio di reazione è raggiunto quando le concentrazioni relative di reagenti e prodotti non cambiano più nel tempo.
Molti enzimi richiedono cofattori per funzionare: ad es. ioni metallici o coenzimi. Se legati covalentemente i cofattori sono detti gruppi prostetici.
Tutti i coenzimi (molecole non proteiche necessarie alla funzione dell’enzima) contengono nella loro molecola una parte vitaminica: il nostro organismo non è in grado di sintetizzare le vitamine, che devono essere assunte con l’alimentazione. Una volta assorbite, le nostre cellule sono in grado di trasformarle nei corrispondenti coenzimi. Data la fondamentale importanza degli enzimi nel consentire il metabolismo si capisce perché la carenza di vitamine coenzimatiche (le vitamine idrosolubili) causi gravi patologie (vedi tabella).
VITAMINA |
COENZIMA |
ENZIMA/FUNZIONE |
SINTOMI DA DEFICIT |
RDA (mg) |
Tiamina (B1) |
TPP |
Piruvato deidrogenasi e KGA-deidrogenasi (ciclo di Krebs) |
Atassia, oftalmoplegia, confusione mentale, faticabilità. |
1-1.5 |
Ac. Pantotenico (B5) |
Coenzima A |
Trasportatore-attivatore di acili |
Non noti, forse perché molto diffuso negli alimenti. |
|
Piridossina (B6) |
PLP |
Transaminasi |
Neuropatie periferiche, anemia sideroblastica. |
1.4-2.0 |
Biotina (H) |
Biocitina |
Carbossilasi |
Prodotta dai batteri intestinali. |
|
Ac. folico |
THF |
Trasf. unità carboniose nella sintesi purine e dTMP (sintesi DNA) |
Anemia megaloblastica; difetti del tubo neurale in neonati da madri deficitarie. |
0.2-0.4 |
Ac. nicotinico (PP) |
NAD+ |
Deidrogenasi (trasf. elettroni) |
Pellagra: dermatite fotoreattiva, diarrea, demenza |
13-19 |
Riboflavina (B2) |
FAD |
Deidrogenasi (trasf. elettroni) |
Cheilite, dermatite, glossite |
1.2-1.7 |
Meccanismi di regolazione dell’attività enzimatica.
Gli enzimi sono i semafori che regolano il traffico di metaboliti lungo le vie metaboliche. L’attività degli enzimi deve essere regolata, in modo da rispondere alle diverse esigenze metaboliche (per es. stato alimentato-digiuno). I meccanismi di regolazione dell’attività enzimatica sono:
Regolazione covalente. Il legame covalente tra enzima e regolatore (tipicamente un fosfato donato dall’ATP) modifica (stimola o inibisce) l’attività enzimatica. Il fosfato è trasferito dall’ATP sull’enzima ad opera di un altro enzima (cinasi, classe transferasi). L’idrolisi del legame enzima-fosfato avviene ad opera di una fosfatasi (classe idrolasi), che ripristina l’attività enzimatica nativa. Cinasi e fosfatasi sono controllate da ormoni: il glucagone stimola le cinasi e l’insulina stimola le fosfatasi (vedi schema)
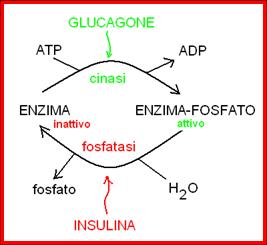
Regolazione allosterica. Il regolatore (attivatore o inibitore), di solito un metabolita oppure ATP o AMP, si lega all’enzima con legami deboli, non covalenti (ponti H, legami ionici) e modifica l’attività enzimatica.
Regolazione ormonale. Gli ormoni possono modificare l’attività degli enzimi sia attraverso meccanismi di regolazione covalente (regolando l’attività di cinasi o fosfatasi che controllano lo stato di fosforilazione degli enzimi), che modificando la velocità di trascrizione dei geni che codificano per alcuni enzimi (induzione o repressione enzimatica).
Produzione di energia metabolica
Il meccanismo generale di produzione di lavoro dall’energia derivante da un flusso elettronico è simile per un motore elettrico e per il nostro organismo.
1- Catabolismo e anabolismo.
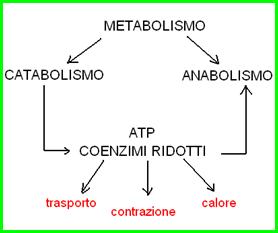
Il metabolismo è l’insieme delle reazioni chimiche che si svolgono nell’organismo. Si divide in catabolismo (reazioni di demolizione, producono energia) e anabolismo (reazioni di sintesi, consumano energia). L’energia prodotta o consumata è sotto forma di coenzimi ridotti (NADH, NADPH e FADH2) e ATP.
Attraverso le reazioni del catabolismo si produce energia, sotto forma di ATP e coenzimi ridotti (NADH, NADPH e FADH2), che viene utilizzata nelle reazioni di biosintesi, per sintetizzare molecole complesse a partire da precursori semplici.
2- Molecole energetiche nella cellula.
L’ATP (adenosine triphosphate) è la moneta corrente per tutte le attività cellulari che producono o consumano energia: ATP viene prodotto nel catabolismo ossidativo e ATP viene speso nelle reazioni biosintetiche o nelle attività cellulari (trasporto di metaboliti e ioni attraverso la membrana, contrazione muscolare, produzione di calore).
L’ATP è energia “pronta” per tutte le esigenze energetiche della cellula.
I coenzimi ridotti NADH e FADH2 che si producono nelle reazioni di ossidazione dei substrati catalizzate dalle ossidoreduttasi sono energia “potenziale”, che sarà trasformata in ATP nella catena respiratoria mitocondriale (3 ATP/NADH e 2 ATP/FADH2).
Il coenzima ridotto NADPH invece è utilizzato come potere riducente nelle biosintesi e NON per la produzione di ATP nella catena respiratoria.
3- Strategie per la produzione di ATP nelle cellule.
Ci sono due modi per produrre ATP attraverso reazioni del catabolismo ossidativo:
- Reazioni di fosforilazione a livello del substrato.
In queste reazioni, catalizzate da cinasi, un intermedio metabolico fosforilato cede il fosfato all’ADP con formazione di ATP secondo lo schema generale:
XP + ADP ® X + ATP
Es. 1,3 bisfosfoglicerato e fosfoenolpiruvato nella glicolisi; creatina fosfato (“riserva di ATP” nel muscolo). Il potenziale di trasferimento del fosfato di questi composti fosforilati è maggiore di quello dell’ATP.
- La fosforilazione ossidativa.
Nel processo della fosforilazione ossidativa (che avviene nei mitocondri) l’energia “potenziale” dei coenzimi ridotti (NADH e FADH2) è trasformata nell’energia “pronta” dell’ATP attraverso un complesso meccanismo. I coenzimi ridotti sono stati prodotti in reazioni di ossidazione dei substrati (glucoso, fruttoso, acidi grassi) nelle varie vie metaboliche del catabolismo (glicolisi, ciclo di Krebs, ossidazione degli acidi grassi).
Pertanto il processo di produzione dell’ATP via fosforilazione ossidativa si può dividere in 2 fasi:
Fase 1 : riduzione dei coenzimi nelle reazioni di ossidazione dei substrati metabolici
Xrid + NAD/FAD ® Xox + NADH/FADH2
Fase 2 : ri-ossidazione dei coenzimi ridotti accoppiata alla produzione di ATP nella fosforilazione ossidativa nei mitocondri
NADH/FADH2 + O2 ® NAD/FAD + H2O
ADP + Pi ® ATP + H2O
La fosforilazione ossidativa è la principale fonte di ATP per la maggior parte delle cellule del nostro organismo.
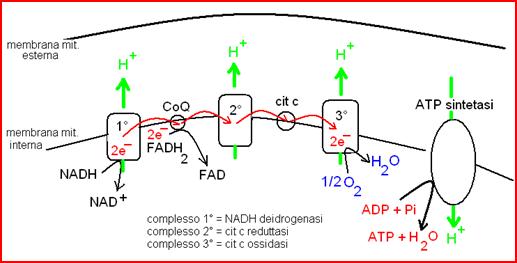
In definitiva, il processo della fosforilazione ossidativa:
- rigenera i coenzimi ossidati (NAD e FAD) necessari nelle reazioni di ossidazione dei substrati metabolici nelle varie vie metaboliche del catabolismo ossidativo
- produce ATP
- produce acqua (l’acqua metabolica, una parte importante del nostro fabbisogno idrico giornaliero)
- produce calore (la proteina termogenina dissipa in parte il gradiente protonico generando calore)
Poiché non tutte le cellule hanno i mitocondri (ad esempio gli eritrociti ne sono privi), nelle cellule prive di mitocondri la produzione di ATP avverrà solo attraverso reazioni di fosforilazione a livello del substrato.
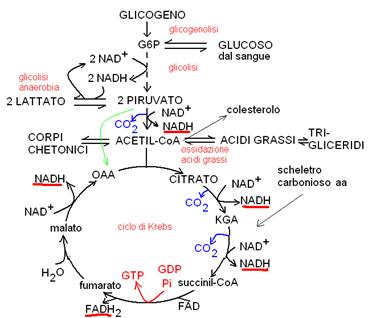
SCHEMA GENERALE DEL CATABOLISMO
Le principali vie metaboliche che prenderemo in esame saranno le seguenti:
Glicogenolisi: demolizione del glicogeno con produzione di glucoso 6-fosfato (G6P).
Glicogenosintesi: sintesi di glicogeno a partire da G6P
Glicolisi: ossidazione del G6P a 2 molecole di piruvato con produzione netta di 2 molecole di ATP.
Glicolisi anaerobia: riduzione del piruvato a lattato per rigenerare il NAD (ossidato) che serve nella glicolisi; quando non ci sono mitocondri oppure l’apporto di ossigeno è insufficiente.
Gluconeogenesi: sintesi di glucosio da precursori non glucidici con consumo di ATP e NADH
Decarbossilazione ossidativa del piruvato ad acetato (acetil coenzima A, acetil CoA): consente l’ingresso del piruvato nel ciclo di Krebs.
Ciclo di Krebs: la principale via ossidativa sulla quale convergono tutti i substrati ossidabili (zuccheri, amminoacidi e acidi grassi); acetil CoA e ossalacetato (OAA) si uniscono a formare citrato, che subisce una serie di reazioni di decarbossilazione ossidativa e di ossidazione con produzione di 3 NADH, 1 FADH2 e 1 ATP per ogni giro del ciclo.
Corpi chetonici: si formano dall’acetato, quando la mancanza di ossalacetato ne impedisce l’ingresso nel ciclo di Krebs (digiuno o diabete).
Ossidazione degli acidi grassi: produce acetil CoA, NADH e FADH2.
Transamminazione degli aminoacidi e deamminazione ossidativa del glutammato: la somma delle due reazioni serve a rimuovere il gruppo amminico dagli amminoacidi, producendo ammoniaca (NH3) e lo scheletro carbonioso dell’amminoacido. L’ammoniaca verrà trasformata in urea nel ciclo dell’urea, mentre lo scheletro carbonioso degli amminoacidi entrerà nel ciclo di Krebs o nella gluconeogenesi (a seconda dell’aa.).
Tutti i coenzimi ridotti (NADH e FADH2) che si producono nelle reazioni di ossidazione dei vari substrati metabolici (glucoso, acidi grassi, acetato) cedono i loro elettroni alla catena di trasporto mitocondriale: al termine del processo della fosforilazione ossidativa, l’ossigeno (trasportato dall’emoglobina a tutti i tessuti) viene ridotto ad acqua e si produce ATP (3 ATP per ogni NADH e 2 ATP per ogni FADH2).
GLICOGENO-LISI E GLICOGENO-SINTESI
Il glucosio è il principale zucchero che fornisce energia metabolica alle cellule di tutti i tessuti, in particolare al cervello. Possediamo “scorte” di glucoso, sotto forma del suo polimero, il glicogeno, nel fegato e nel muscolo. Una molecola di glicogeno contiene molte migliaia di molecole di glucosio unite le une alle altre in una catena ramificata. Il glicogeno epatico è a disposizione di tutti i tessuti, grazie alla presenza nel fegato di un enzima (glucosio 6-fosfatasi) che catalizza la defosforilazione del glucosio-6-fosfato, prodotto dalla demolizione del glicogeno, a glucoso. Il glucosio libero può uscire dalle cellule epatiche e raggiungere il sangue. Invece il glicogeno muscolare è solo ad uso e consumo del muscolo perché il glucoso 6-fosfato che si produce dalla demolizione del glicogeno muscolare non può essere defosforilato e quindi non può uscire dalla cellula muscolare: viene ossidato nella glicolisi per produrre ATP nel muscolo stesso.
La glicogenolisi è la demolizione del glicogeno con produzione di glucoso 6-fosfato.
La glicogenosintesi è la via metabolica che produce glicogeno a partire da glucosio-6-fosfato con consumo di ATP.
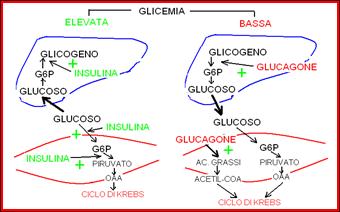
Regolazione. Insulina e glucagone sono due ormoni proteici prodotti dalle cellule b (insulina) e a (glucagone) del pancreas endocrino e sono i principali responsabili della regolazione della sintesi e della demolizione del glucagone nel fegato.
Il glucagone attiva nel fegato la demolizione del glicogeno e inibisce la sintesi. L’insulina attiva la sintesi del glicogeno e inibisce la demolizione.
Il rapporto glucagone/insulina nel sangue è controllato dalla glicemia:
glicemia elevata ® basso rapporto glucagone/insulina
glicemia bassa ® alto rapporto glucagone/insulina
GLICOLISI
La glicolisi è la via metabolica che ossida il glucoso a piruvato (2 molecole) con produzione di 2 molecole di ATP e 2 molecole di NADH. Presente in tutti i tessuti, può funzionare anche in assenza (o carenza) di ossigeno: glicolisi anaerobia (eritrociti, muscolo in contrazione). Il glucoso (concentrazione nel sangue 80-100 mg/100ml) entra nelle cellule grazie ad un trasportatore proteico presente sulla membrana di tutte le cellule. L'insulina stimola il trasporto del glucoso in tutti i tessuti, tranne fegato, cervello e globuli rossi.
Una volta entrato nelle cellule il glucoso viene fosforilato a spese di ATP: il glucoso 6-fosfato non può più uscire dalle cellule e viene ossidato nella glicolisi con produzione di 2 molecole di piruvato, 2 molecole di NADH (che possono rendere 6 ATP totali nella fosforilazione ossidativa), e 2 molecole di ATP (che si produce direttamente in reazioni di fosforilazione a livello del substrato).
La glicolisi è attivata da una bassa carica energetica e dall'insulina; è inibita da una alta carica energetica e dal glucagone.

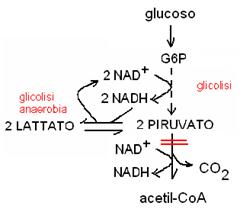
Glicolisi anaerobia.
In presenza di mitocondri e di ossigeno il piruvato viene ossidato nel ciclo di Krebs con produzione di ATP e coenzimi ridotti e il NADH (2 molecole/glucoso) viene riossidato nella catena respiratoria. In mancanza di mitocondri (negli eritrociti) o quando l'apporto di ossigeno col sangue è insufficiente alle necessità metaboliche (muscolo in prolungata contrazione) il NADH non può essere riossidato nella catena respiratoria alla stessa velocità alla quale è prodotto nella glicolisi. Perché la glicolisi possa continuare è necessario rigenerare il NAD (ossidato) attraverso la reazione di riduzione del piruvato a lattato, catalizzata dall’enzima lattico deidrogenasi (LDH):
piruvato + NADH + H+ « lattato + NAD
Il lattato è un prodotto di “scarto” della glicolisi, che esce dalle cellule che lo hanno prodotto (eritrociti, muscolo in contrazione) e raggiunge con il sangue il fegato, dove verrà trasformato in nuovo glucoso con la gluconeogenesi.
La riduzione del piruvato a lattato ad opera dell’enzima LDH e del NADH serve a rigenerare il NAD ossidato (NAD+) che serve nella reazione della gliceraldeide 3-fosfato deidrogenasi nella glicolisi. In questo modo, la glicolisi può continuare anche se non funziona la catena respiratoria.
GLUCONEOGENESI
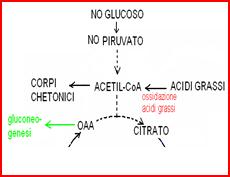
La gluconeogenesi è la sintesi di nuovo glucoso a partire da precursori non carboidrati (lattato, piruvato, alcuni amminoacidi). Avviene solo nel fegato. La sintesi di una molecola di glucoso a partire ad esempio da 2 molecole di piruvato “costa” 6 ATP e 2 NADH. Quando il fegato è impegnato nella gluconeogenesi “consuma” ossalacetato, che è un intermedio di questa via biosintetica. Quindi una gluconeogenesi molto attiva può causare carenza di ossalacetato e arresto del ciclo di Krebs. Questo si verifica ad esempio nel digiuno prolungato. L’arresto del ciclo di Krebs impedisce l’ossidazione dell’acetil CoA con formazione dei corpi chetonici.
Regolazione. Gli enzimi che catalizzano le reazioni irreversibili della gluconeogenesi sono tutti attivati dall’ormone pancreatico glucagone, e inibiti dall’insulina. Quindi, una diminuzione della glicemia induce rilascio di glucagone dalle cellule a-pancreatiche ed il glucagone stimola la gluconeogenesi epatica, con conseguente ripristino di normali livelli glicemici:
Ipoglicemia ® glucagone ® attivazione gluconeogenesi epatica ® glicemia risale
CICLO DI KREBS
Il ciclo di Krebs è la via finale comune del catabolismo glucidico (via acetil CoA), proteico (via alcuni intermedi del ciclo di Krebs) e lipidico (via acetil CoA).
Per ogni acetil CoA ossidato nel ciclo di Krebs si producono 3 NADH, 1 FADH2 e 1 ATP: nella fosforilazione ossidativa ogni NADH consente la produzione di 3 ATP e ogni FADH2 consente la produzione di 2 ATP. Quindi un giro del ciclo di Krebs “rende” 12 ATP. Infine, si producono anche molecole di CO2 attraverso reazioni di decarbossilazione ossidativa: è questa la CO2 che espiriamo!
Il ciclo avviene nei mitocondri: il piruvato prodotto dalla glicolisi (nel citoplasma) entra nel mitocondrio. Una volta nel mitocondrio, il piruvato subisce una reazione di decarbossilazione ossidativa (nel ciclo di Krebs ne troviamo altre due simili) che lo trasforma in acetil CoA:
Piruvato + NAD + coenzima A ® acetil CoA + NADH + H+ + CO2
Nella prima reazione del ciclo, l’acetil CoA condensa con ossalacetato e si forma citrato (6 carboni). Le successive reazioni del ciclo di Krebs comprendono reazioni di decarbossilazione ossidativa (con produzione di 2 molecole di CO2 per ogni “giro” del ciclo) e reazioni di ossidazione senza decarbossilazione. Alla fine di un giro del ciclo si rigenera una molecola di ossalacetato e si producono 3 NADH, 1 FAH2 e 1 ATP (vedi schema riassuntivo vie cataboliche).
Regolazione. Le deidrogenasi che catalizzano le reazioni di decarbossilazione ossidativa (piruvato deidrogenasi, isocitrato deidrogenasi e a-chetoglutarato deidrogenasi), che sono tutte reazioni irreversibili, sono inibite allostericamente da ATP e da NADH (alta carica energetica).
Il ciclo di Krebs “gira” finchè c’è tanto OAA quanto acetil CoA: se la [acetil CoA] è maggiore della [OAA], l’acetil CoA in eccesso viene utilizzato nel fegato per la sintesi dei corpi chetonici (acetoacetato, idrossibutirrato e acetone). Questa situazione si verifica nell’ipoglicemia intracellulare (causata da digiuno prolungato o da diabete di tipo I). In queste condizioni l’OAA viene dirottato verso la gluconeogenesi, stimolata dal glucagone, e contemporaneamente aumenta la produzione di acetil CoA dalla demolizione degli acidi grassi, che viene stimolata dal glucagone per far fronte alla carenza energetica.
I corpi chetonici sono molecole acide (liberano H+ in soluzione) e, se presenti in elevata concentrazione nel sangue, provocano acidosi (pH del sangue < 7,3), detta “metabolica” per distinguerla dall’acidosi “respiratoria”, causata invece da accumulo di CO2 nel sangue per insufficiente ventilazione polmonare. L’acidosi, come l’alcalosi (pH del sangue >7,3), sono condizioni molto pericolose: una variazione del pH del sangue anche di 1 decimo di unità di pH può determinare la morte per insufficienza funzionale di tutti i tessuti (gli enzimi all’interno delle cellule non funzionano più).
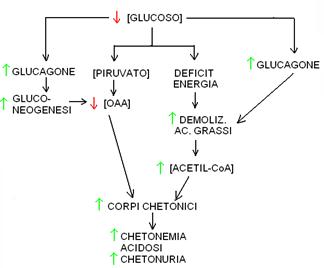
Condizioni metaboliche che determinano chetogenesi epatica.
OSSIDAZIONE DEGLI ACIDI GRASSI
I trigliceridi (glicerolo esterificato con 3 acidi carbossilici a lunga catena, detti acidi grassi) sono la principale scorta energetica del nostro organismo depositata nel tessuto adiposo, come dimostrato dal bilancio delle kcal totali presenti nel nostro organismo sotto forma di molecole ossidabili:
trigliceridi 125000 kcal
proteine mobilizzabili 25000 kcal
glicogeno (muscolo) 1200 kcal
glicogeno (fegato) 600 kcal
glucoso (sangue) 40 kcal
I legami estere che uniscono gli acidi grassi al glicerolo sono scissi per introduzione di acqua (idrolisi) da enzimi detti lipasi, attivati dal glucagone.
Gli acidi grassi liberati nel tessuto adiposo entrano nel torrente circolatorio, dove sono veicolati dall’albumina a tutti i tessuti. Poiché sono molecole lipofile attraversano liberamente la membrana cellulare. L’ossidazione degli acidi grassi avviene nei mitocondri di tutti i tessuti e produce acetil CoA e coenzimi ridotti (NADH e FADH2). Il bilancio energetico è di 2 ATP consumati (per l’attivazione) e 131 ATP prodotti per ogni acido palmitico (16 atomi di carbonio) ossidato.
Regolazione. Il glucagone, prodotto dal pancreas in condizioni di ipoglicemia, stimola la liberazione degli acidi grassi dai trigliceridi e la loro ossidazione in tutti i tessuti. Il glucagone stimola anche la gluconeogenesi epatica, che consuma OAA: per questo motivo, nel fegato, si può verificare uno squilibrio tra la concentrazione di OAA (bassa) e quella di acetil CoA (alta, perché continuamente prodotto dalla ossidazione degli acidi grassi). Questa condizione porta all’accumulo nel fegato dei corpi chetonici. Trasportati attraverso il sangue questi giungono al rene, vengono filtrati dal glomerulo e si ritrovano nelle urine (chetonuria).
DEGRADAZIONE DEGLI AMMINOACIDI
Gli amminoacidi contengono un gruppo amminico (-NH2), che non è presente in alcun metabolita intermedio delle vie metaboliche (glicolisi, gluconeogenesi, ossidazione e sintesi degli acidi grassi, ciclo di Krebs). Perché gli aa. possano entrare nel metabolismo energetico è necessario che il gruppo amminico venga rimosso. Lo scheletro carbonioso degli aa. entra allora nel ciclo di Krebs o nella gluconeogenesi, mentre l’ammoniaca viene trasformata in urea nel fegato.
- Transaminazione e deaminazione ossidativa.
Dei 20 amminoacidi che costituiscono le proteine, l’unico che può essere deaminato (distacco del gruppo amminico) direttamente è il glutammato, con formazione di ammoniaca (NH3) e a-chetoglutarato (KGA), che è un intermedio del ciclo di Krebs. La reazione è reversibile ed è catalizzata dall’enzima glutammico deidrogenasi; questa reazione avviene in tutti i tessuti.
Glutammato + NAD + Pi « a-chetoglutarato + NADH + NH3
Glutammico deidrogenasi
Come fanno gli altri aa. a perdere il gruppo amminico?
Il meccanismo è il seguente: in una reazione di transaminazione il gruppo amminico dell’amminoacido 1 viene trasferito sull’a-chetoglutarato. L’amminoacido 1 diventa un a-chetoacido e l’a-chetoglutarato diventa glutammato (con il gruppo amminico dell’amminoacido 1).
amminoacido 1 + a-chetoglutarato « chetoacido 1 + glutammato
transaminasi
Le transaminasi sono una famiglia di enzimi presenti in tutti i tessuti, ma particolarmente abbondanti nel fegato e nel muscolo.
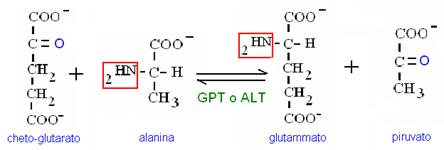
AST e ALT sono enzimi di interesse diagnostico (epatite, cirrosi, infarto miocardio), perché si ritrovano in quantità elevata nel siero a seguito di un danno cellulare epatico o muscolare. Inoltre, poiché la AST è un enzima mitocondriale mentre la ALT è citosolica, un danno cellulare lieve libererà attività ALT mentre un danno cellulare grave (con rottura anche dei mitocondri) libererà anche attività AST. Dal rapporto AST/ALT nel siero si ricavano quindi anche informazioni sulla gravità del danno cellulare.
Qual’è il destino dell’ammoniaca (NH3) ?
L’ammoniaca è tossica, perché quando presente in elevata quantità nel sangue entra nelle cellule e sposta verso sinistra l’equilibrio della reazione della glutammico deidrogenasi (vedi sopra) con consumo di a-KGA, che è un intermedio del ciclo di Krebs. Se viene consumato tutto l’a-KGA (che diventa Glu), il ciclo di Krebs si arresta e non si produce più ATP nella cellula. Il tessuto più sensibile al danno da NH3 è il cervello (coma da iperammoniemia).
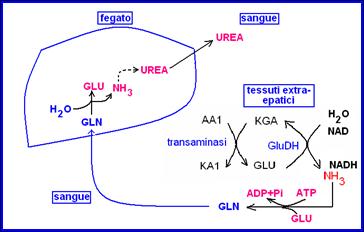
L’ammoniaca prodotta dalla deaminazione del glutammato (che avviene in tutti i tessuti) viene trasportata nel sangue sotto forma di glutammina e raggiunge il fegato, dove l’ammoniaca viene trasformata in urea con dispendio di 4 ATP per molecola di urea prodotta.
L’urea prodotta nel fegato raggiunge con il sangue il rene. La concentrazione ematica dell’urea (20 mg/100 ml) è 100 volte maggiore di quella dell’NH3. L’urea viene filtrata dal glomerulo, non è riassorbita dal tubulo e quindi viene escreta nelle urine (circa 20g/die).
CORRELAZIONI METABOLICHE TRA ORGANI
Fegato e omeostasi glicemica.
Il fegato è responsabile del mantenimento di una glicemia costante durante il giorno, nonostante l’apporto saltuario di glucidi con l’alimentazione.
gluconeogenesi

Il fegato produce glucosio (con la gluconeogenesi) e lo esporta nel sangue ad uso e consumo degli altri organi (primo fra tutti il cervello). La gluconeogenesi avviene a partire da alcuni amminoacidi, glicerolo, piruvato e da acido lattico. Il lattato è il prodotto “di scarto” del muscolo e degli eritrociti, che lo producono nella glicolisi anaerobia: attraverso il sangue il lattato giunge al fegato che lo utilizza per fare nuovo glucoso (ciclo del lattato tra muscolo e fegato). L’enzima glucoso 6-fosfatasi defosforila il glucoso 6-fosfato, alla fine della gluconeogenesi, ed il glucoso libero passa nel sangue e viene utilizzato dal muscolo nella glicolisi.
Il metabolismo del glucoso e del glicogeno nel fegato è regolato dagli ormoni pancreatici insulina e glucagone. Il glucagone (rilasciato dal pancreas endocrino in condizioni di ipoglicemia) stimola la gluconeogenesi e la glicogenolisi nel fegato: il glucoso 6-fosfato prodotto dal glicogeno viene defosforilato dalla glucoso 6-fosfato fosfatasi (enzima solo epatico!) e il glucoso libero può uscire dalle cellule epatiche e raggiungere con il sangue tutti i tessuti. L’insulina (rilasciata dal pancreas endocrino in condizioni di glicemia elevata) stimola la glicogenosintesi.
Il fegato è anche il deposito di glicogeno che viene demolito nei periodi di ipoglicemia per rifornire di glucoso il sangue e viene sintetizzato nei periodi di iperglicemia per mettere da parte la riserva di glucoso.
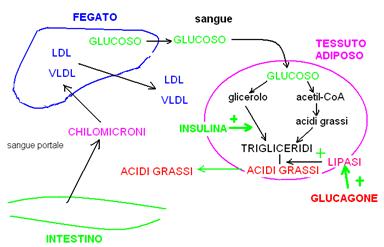
Fegato e metabolismo lipidico.
Lipoproteine.
Il tessuto adiposo assorbe dal sangue le lipoproteine prodotte dal fegato e ne utilizza i componenti lipidici per accumulare trigliceridi negli adipociti.
L’insulina (che segnala abbondanza di glucoso nel sangue) stimola la sintesi dei trigliceridi negli adipociti. In caso di ipoglicemia, il glucagone stimola le lipasi che idrolizzano i trigliceridi a glicerolo e acidi grassi. Questi ultimi attraverso il sangue raggiungono i tessuti e vengono ossidati per fornire energia al posto dei glucidi.
Con i grassi che arrivano dal circolo portale (cioè di origine alimentare, assorbiti dall’intestino) il fegato produce ed esporta nel sangue le lipoproteine (LDL, HDL e VLDL) che sono il veicolo attraverso cui i grassi (trigliceridi e colesterolo) viaggiano nel sangue e raggiungono i vari tessuti. Il guscio proteico assicura la solubilità in acqua dei lipidi e ne consente la captazione da parte dei tessuti attraverso recettori specifici.
Chetogenesi.
In questo scenario metabolico (ipoglicemia, alto rapporto glucagone/insulina nel sangue) il fegato è impegnato nella glicogenolisi e nella gluconeogenesi, per rifornire di glucoso il sangue. La gluconeogenesi consuma ossalacetato, che è anche necessario nel ciclo di Krebs. Di conseguenza si verifica una carenza relativa di ossalacetato (rispetto all’acetil CoA) nel ciclo di Krebs (solo a livello epatico!). L’acetil CoA in eccesso viene convertito in corpi chetonici (acetoacetato, idrossibutirrato e acetone) che escono dal fegato e attraverso il sangue raggiungono i tessuti. Il muscolo (cardiaco e scheletrico) è il principale utilizzatore dei corpi chetonici prodotti dal fegato: le scorte muscolari di glicogeno (ad uso e consumo solo del muscolo e quindi di più lunga durata rispetto a quelle epatiche) ed il continuo apporto di glucoso col sangue (gluconeogenesi epatica) fanno sì che il muscolo, contrariamente al fegato, abbia ossalacetato a disposizione per ossidare l’acetil CoA nel ciclo di Krebs. Se però la sintesi di corpi chetonici nel fegato supera la capacità di utilizzazione del muscolo, i corpi chetonici si accumulano nel sangue (chetonemia), vengono filtrati dal glomerulo, non riassorbiti dal tubulo e finiscono nelle urine (chetonuria), dove normalmente sono assenti.
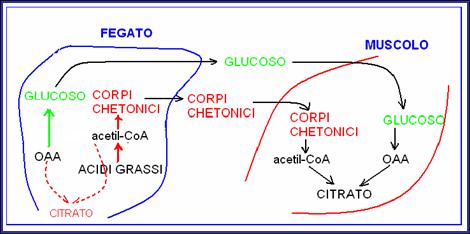
Chetogenesi epatica e utilizzazione muscolare dei corpi chetonici in condizioni di ipoglicemia.
N.B: Il muscolo possiede anche scorte proprie di glucoso (glicogeno) da cui produrre OAA!
In definitiva, la presenza di chetonemia elevata e chetonuria denuncia uno squilibrio tra catabolismo degli zuccheri (poco) e catabolismo dei grassi (tanto). E’ una condizione che si verifica nel digiuno prolungato e nel diabete insulino-dipendente (tipo I) non controllato (dalla terapia insulinica).
L’accumulo di corpi chetonici nel sangue causa acidosi (detta “metabolica” per distinguerla da quella respiratoria, causata da una insufficiente ventilazione e dal conseguente accumulo di CO2 nel sangue) ed è una condizione potenzialmente molto pericolosa. La terapia dipende dalla condizione metabolica che ha determinato la chetogenesi e consiste nella somministrazione di glucoso (nel digiuno) o nella infusione di insulina (nel diabete tipo I): confondere le due situazioni metaboliche può avere esito mortale!
Il diabete di tipo I (insulino-dipendente) è una malattia metabolica grave causata dalla incapacità dei tessuti di utilizzare metabolicamente il glucoso, perché manca l’insulina, che normalmente stimola la glicogenosintesi epatica, il trasporto del glucoso dentro le cellule dei tessuti – tranne fegato e cervello! – e la glicolisi in tutti i tessuti. Il risultato è un quadro sintomatologico grave.
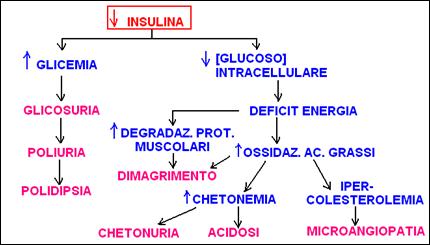
Il deficit di insulina (provocata, si ritiene, da una aggressione autoimmune ai danni delle cellule b-pancreatiche) riduce il trasporto del glucoso dentro le cellule dei tessuti (tranne fegato e cervello, che non richiedono insulina per il trasporto del glucoso) e riduce la capacità dei tessuti di utilizzare metabolicamente il glucoso. Il risultato è una condizione di ipoglicemia intracellulare, che contrasta paradossalmente con la iperglicemia extracellulare (nel sangue).
L’iperglicemia causa glicosuria (glucoso nelle urine, normalmente assente) perché il tubulo renale non è in grado di riassorbire tutto il glucoso filtrato dal glomerulo. L’elevata concentrazione di glucoso nel filtrato glomerulare richiama acqua nel tubulo renale (per osmosi) e aumenta il volume dell’escrezione urinaria (poliuria) fino a 10 volte il normale. La disidratazione che ne deriverebbe è impedita solo dalla aumentata introduzione di liquidi (polidipsia), indotta dal senso di sete. [Il diabetico beve tanto perché fa tanta pìpì, non viceversa!]
Il deficit energetico causato dall’assenza della fonte glucidica induce un aumento (rispetto al normale) dell’ossidazione di acidi grassi (dai trigliceridi) ed amminoacidi (dalle proteine) per produrre energia metabolica (dimagrimento). La mancanza di glucoso (e quindi di OAA) impedisce la completa ossidazione dei prodotti del catabolismo lipidico e amminoacidico (acetil CoA e intermedi del ciclo di Krebs) nel ciclo di Krebs: il fegato produce ed esporta nel sangue corpi chetonici e colesterolo (anch’esso sintetizzato a partire da acetil CoA).
L’incapacità dei tessuti extraepatici di utilizzare i corpi chetonici (perché manca glucoso e quindi OAA) determina l’accumulo dei copri chetonici nel sangue (chetonemia e acidosi metabolica) e quindi nelle urine (chetonuria).
Infine l’aumentata [colesterolo] nel sangue, forse la conseguenza più perniciosa dell’aumentata disponibilità di acetil CoA nel fegato, causa una precoce aterosclerosi, responsabile della progressiva ostruzione dei piccoli vasi (microangiopatia diabetica) che negli anni causa l’insufficiente ossigenazione di molti tessuti (reni, cuore, retina, microcircolo delle estremità).
Il diabete di tipo I è una malattia gravemente invalidante. La diagnosi avviene in età pediatrica e, se controllato con una precoce e bilanciata terapia insulinica (insulina ricombinante umana) e con la dieta e l’esercizio fisico è compatibile con una vita normale.
Il diabete di tipo II è invece causato da una ridotta sensibilità dei tessuti all’insulina, che è prodotta dal pancreas in quantità normale. Il problema quindi riguarda il recettore e non l’ormone. Mentre il diabete tipo I ha tipicamente esordio in età giovanile e comporta dimagrimento (vedi sopra), il diabete tipo II ha esordio nell’età adulta/avanzata ed è associato a sovrapeso. La tabella riassume le principali caratterisitiche del diabete tipo I e tipo II.
Diabete |
Patogenesi |
Manifestaz. cliniche |
Terapia |
Insulino- |
Distruzione autoimmune cellule b (autoanticorpi anti-insulina o cell. b nel 95% dei casi) |
Poliuria |
Insulina, dieta, attività |
Insulino- |
Insulino-resistenza associata |
Asintomatico, esordio |
Ipoglicemizzanti orali |
Diabete in gravidanza. Il diabete gestazionale è una forma di diabete che insorge in gravidanza e termina, generalmente, con la fine di questa. E’ una fra le complicanze più frequenti della gravidanza (2-5%), seconda solo all'ipertensione (5-10%). Una delle principali cause del diabete gestazionale è la produzione di ormoni (Lattogeno Placentare, cortisolo e prolattina) che interferiscono con l'azione dell'insulina. In donne predisposte questa interferenza è molto accentuata e determina l'insorgenza del diabete gestazionale. Sviluppare un diabete gestazionale può essere un fattore di rischio per sviluppare negli anni un diabete di tipo II.
Fegato e organicazione dell’ammoniaca.
Il fegato è l’unico organo che può trasformare l’ammoniaca derivante dal catabolismo degli amminoacidi (ma non solo!) in urea. L’ammoniaca prodotta nei vari tessuti dalle reazioni di transamminazione e deamminazione ossidativa del glutammato viene legata al glutammato, con formazione di glutammina, dall’enzima glutammina sintetasi (con spesa di ATP).
La glutammina raggiunge il fegato attraverso il sangue e nel fegato l’enzima glutamminasi libera l’ammoniaca (NH3).
Quindi NH3 non viaggia nel sangue come tale, ma sotto forma dell’innocuo amminoacido glutammina.
Concentrazioni ematiche di alcune importanti biomolecole.
Glucoso 80-100 mg/100 ml (5 mM)
Urea 20 mg/100 ml (3 mM)
Acido urico 3-7 mg/100 ml
Ammoniaca 60 mg/100 ml (30 mM)
Trigliceridi 70-140 mg/100 ml
Colesterolo 150-200 mg/100 ml
Corpi chetonici 1,5 mg/100 ml (0,2 mM)
Digiuno 3-5 mg/100 ml
Diabete non controllato fino a 150 mg/100 ml (20 mM)
http://www.medicina.unige.it/didattica/percorsiFormativi/EdProf2008-09/materiale/APPUNTI%20DI%20BIOCHIMICA%20PFS%20Educazione%20professionale.doc
Biochimica
Prof. Solinas
Lezione 14/10/04
Tutti i lucidi delle lezioni e tutte le 220 possibili domande d’esame sono disponibili all’Edisu. L’esame sarà scritto, composto da 30 quesiti estratti dai 220, a scelta multipla di 4 risposte di cui una o più sono giuste. Al termine di ogni lezione verranno proposti dei quesiti fino a vederli tutti e 220.
La Biochimica è la scienza che studia la chimica della materia vivente e della vita cellulare.
- Struttura delle proteine (in particolare dell’Emoglobina che si farà a fondo perché è responsabile del trasporto dell’ossigeno).
- Struttura dei carboidrati.
- Struttura dei lipidi.
- Processi metabolici.
Le molecole che studia la Biochimica hanno grandezze dell’ordine di 10-9m = 1 nm (manometro) = 1 miliardesimo di metro.
Le molecole organiche presenti nelle cellule sono formate in gran parte dai seguenti elementi: C, N, O, Na, Mg, P, S, Cl, K, Ca. La materia vivente differisce dal mondo inanimato non tanto nella composizione ma dal fatto che essa è costituita dagli stessi elementi ma organizzati in macrostrutture che reagiscono e si scambiano informazioni tra loro.
La cellula è costituita da polisaccaridi, proteine, lipidi e acidi nucleici. Il DNA si trova solo nel nucleo. Le proteine, i lipidi e i polisaccaridi si trovano in proteine, membrane, citoplasma e organuli.
La materia vivente è costituita per il 50% da acqua ed è in essa che si svolgono tutte le reazioni chimiche. L’acqua è formata da un legame covalente tra due atomi di H ed uno di O. Questi atomi non sono disposti in linea ma a triangolo. L’angolo che l’O forma con i due atomi di H è di circa 105°.
Legame covalente: due atomi mettono a disposizione degli elettroni che vengono condivisi con un altro atomo. Ciascuno dei due atomi di idrogeno condivide il suo elettrone con l’atomo di ossigeno.
Legame ionico: gli elettroni si dispongono tutti su uno dei due atomi.
Il legame all’interno della molecola d’acqua è molto forte. Per separarne gli elementi occorrono energie molto forti come le radiazioni ionizzanti o una temperatura > 1.600°C. La lunghezza del legame covalente è di circa 0,9Å (1 Ångstrom = 0,1 nm). Il raggio di Van der Waals è la distanza dall’atomo entro la quale si verificano fenomeni di repulsione o attrazione di altre molecole.
L’ossigeno ha la caratteristica di essere elettronegativo (attira a sé gli elettroni) più dell’Idrogeno, quindi, nella molecola d’acqua, l’ossigeno attirerà a sé gli elettroni condivisi per un tempo maggiore dell’idrogeno. Per questo motivo la molecola d’acqua è un dipolo, ha cioè un polo negativo (ossigeno) ed uno positivo (idrogeno). Questa caratteristica consente alle molecole d’acqua di formare dei “legami a idrogeno” in cui l’atomo di idrogeno fa da ponte di legame tra due atomi d’ossigeno formando una rete di 4 molecole d’acqua (clatrato). Qualsiasi sostanza solubile in acqua (sale da cucina) va ad interagire con i legami a idrogeno sui poli della molecola d’acqua. L’olio non si sciogle (interagisce) in acqua perché è apolare. Se si fa bollire l’acqua si interrompono i legami a idrogeno tra le molecole d’acqua, non quelli all’interno della molecola.
Ionizzazione delle molecole:
L’acqua pura non conduce elettricità. Se all’acqua si aggiunge un soluto (come il sale da cucina) conduce elettricità. NaCl sciolto in acqua forma Na+ Cl-. NaCl è un elettrolita forte perché in acqua si dissocia completamente. NaCl -> Na+ Cl-. Un acido debole si comporta invece nel modo seguente:
AH<->A+ + H+: non tutte le molecole di AH si dissociano ma solo una parte. Più una sostanza è debole meno si dissocia.
K è in sostanza il rapporto tra la parte di AH che si dissocia e quella che non lo fa. Più K è grande più siamo in presenza di elettroliti forti.
CH3COOH (acido acetico) <-> CH3COO- + H+ K=1,73*10-5
H2CO3 (acido carbonico) <-> H+ + HCO3- K=3,5*10-7
HCO3- a sua volta <-> H+ + CO3- K2=4,5*10-11
Siccome 10-5 è un numero più grande di 10-7, l’acido acetico è più acido (dissociante) dell’acido carbonico.
L’acido è una sostanza AH che rilascia ioni H+ in acqua. L’acqua viene così “protonata”.
HCl è un acido molto forte perché ionizza completamente: HCl + H2O -> Cl- + H3O+ (ione idronio).
Tanto più l’acido è forte, tanto più “protona” l’acqua. Anche le molecole d’acqua possono protonarsi tra di loro.
HOH + HOH <-> OH- + H3O+. Quindi l’acqua è un debole elettrolita perché dissocia (poco) in ioni.
a 24°C solo 18g su 1 tonnellata.
[H+]*[OH-]=10-14 ma [H+] esiste nella stessa quantità di [OH-] (uno ione ciascuno per molecola dissociata, quindi
[H+]*[H+]=10-14 quindi
[H+]2=10-14 quindi
[H+]=10-7, infatti l’acqua ha un pH di 7.
Ma [H+] era uguale a [OH-], quindi anche gli ioni di [OH-] sono presenti nella misura di 10-7.
In un l di acqua pura ci sono 10-7 ioni [H+] e 10-7 ioni [OH-]
[OH-] = ioni ossidrili.
Se all’acqua si aggiunge un acido ([H+]) o una base ([OH-]) si avrà la prevalenza di uno dei due.
- Soluzione acida se concentrazione idrogenionica > 10-7 (es. 10-6)
- Soluzione basica se concentrazione idrogenionica < 10-7 (es. 10-8)
- Soluzione neutra se concentrazione idrogenionica = 10-7
Il pH = all’esponente di 10 * -1
Il pH fisiologico del sangue è 7,4. Si parla di acidosi per valori inferiori (7,2 è ancora tecnicamente una soluzione basica ma è acida rispetto al pH fisiologico) e di alcalosi per valori > 7,4. In condizioni estreme, il pH può variare di ± 0,5 ma, ai valori estremi, si è in condizioni “da rianimazione”. Le variazioni fisiologiche sono nell’ordine di ± 0,03.
Nell’organismo il pH è fortemente controllato dai sistemi tampone. Il sistema più efficace è quello della CO2. Il pH deve essere costante perché le strutture di proteine ed enzimi sono fortemente influenzate dalle variazioni di pH.
Un acido produce ioni H+ (protoni) se disciolto in acqua.
- Acidi minerali (forti):
- HCl (acido cloridrico, l’unico presente nell’organismo ma “isolato” nello stomaco” -> H+ + Cl-.
- HNO3 -> H+ + NO3-
- H2SO4 -> H+ + HSO4-
- Acidi deboli:
- H3PO4 <-> H+ + H2PO4-
- H2CO3 <-> H+ + HCO3-
- CH3COOH <-> H+ + CH3COO-
Una base invece produce OH- se disciolta in acqua.
- NaOH -> Na+ + OH-
- KOH -> K+ + OH-
- NH4OH -> NH4+ + OH-
Un acido + una base produce un sale + acqua:
- AH + BOH <-> AB (sale) + H2O
- HCl + NaOH <-> NaCl + H2O
Acqua pura e saliva hanno un pH attorno a 7.
Il latte ha un pH attorno a 6,5.
Il succo d’arancia e la Coca Cola hanno un pH attorno a 4,5
I succhi gastrici hanno un pH attorno a 2.
Un detersivo domestico ha un pH attorno a 11,5.
Un acido cede protoni, una base li acquista:
H2SO4 (acido) + H2O (base) <-> HSO4- + H3O+. L’acqua quindi può comportarsi da base.
Per essere un acido una molecola deve contenere H e il suo grado di acidità dipende dagli atomi a cui è legato.
Sistemi arteriosi del sangue
Nel corso delle attività cellulari sono prodotti acidi e basi:
- H2CO3 acido carbonico
- CH3CHOHCOOH acido lattico
- CH3CH2COOH acido acetico
- NH3 (ammoniaca, base)
- R-NH2 (ammine, basi)
Che provocherebbero variazioni di pH nel sangue. Come reagisce l’organismo?
- Meccanismi fisiologici: i reni possono secernere o ritenere sostanze di azione contraria a quelle che hanno determinato la variazione.
- Meccanismi biochimici:
- Tampone acido carbonico-bicarbonato
H2CO3 – HCO3-
La CO2 è un gas parzialmente solubile in acqua e si trova, nell’organismo, in equilibrio tra la sua forma gassosa (CO2g) e quella acquosa (CO2H20) prodotta dal metabolismo cellulare.
CO2H20 reagisce con l’acqua formando H2CO3 (acido carbonico) in presenza dell’enzima anidrasi carbonica.
H2CO3 <-> H+ + HCO3-
In sintesi:
CO2g <-> CO2H2O + H2O <-> H2CO3 <-> H+ + HCO3-
Il tutto in equilibrio continuo. Se varia il pH viene prodotta una quantità più o meno grande di CO2 che viene eliminata o ritenuta regolando la ventilazione polmonare. Questo è in parte il motivo dell’aumento della frequenza respiratoria durante l’esercizio fisico che produce acido lattico, che per il sistema del tampone, si riflette su di una maggiore quantità di CO2 (l’altro motivo dell’aumento della ventilazione è l’aumentato fabbisogno di ossigeno). Se il sangue diventa alcalino diminuisce la frequenza respiratoria.
- Tampone proteico
COOH
COOH
COOH
COOH
COO-
COO-
COO-
COO-
H+---à
ß---OH-
Le proteine si comportano come acidi estremamente debole in equilibrio con i propri Sali formando un sistema proteine – proteinati provvisto di tampone.
- Tampone fosfato
H2PO4 <-> H+ + HPO42-
(fosfato monobasico) (fosfato bibasico)
Anche questi sono normalmente presenti nel sangue.
Lezione del 21/10/04 saltata
Lezione del 28/10/04 saltata
Lezione del 04/11/04
Le lezioni saltate verranno recuperate dopo le vacanze.
La chimica del carbonio (chimica organica)
I composto organici sono quelli in cui l’atomo essenziale è rappresentato dal carbonio. Gli idrocarburi sono i più semplici, formati solo da atomi di carbonio e idrogeno. L’atomo di carbonio può formare 4 legami (tetravalente). Il metano:
è la molecola organica più semplice. Gli atomi di carbonio e di idrogeno condividono un elettrone ciascuno formando legami con due elettroni condivisi (legame covalente). CH4 è la “formula bruta” del metano. A livello tridimensionale, la forma è tetraedrica con l’atomo di carbonio al centro e quelli di idrogeno ai 4 angoli del tetraedro. Il carbonio e l’idrogeno hanno la stessa elettronegatività, pertanto, gli elettroni sono equamente condivisi tra i due atomi e non permangono nella sfera d’influenza di un elemento più a lungo che nell’altro. Ad esempio, l’ossigeno è molto più elettronegativo dell’idrogeno e quindi nell’acqua gli elettroni condivisi sono più vicini e rimangono per più tempo vicini all’atomo di ossigeno che a quello di idrogeno.
Il legame ionico non è un legame di condivisione o incastro, è solo una reciproca attrazione elettrica di due elementi. I legami ionici si spezzano molto facilemente, per quanto riguarda il sale da cucina Na+Cl-, basta immergerlo in acqua per separare i due elementi e creare atomi di Na+ e Cl-
Gli idorocarburi si dividono in:
- alifatici:
- alcani CnH2n+2:
- Etano: se, al metano si toglie un atomo di idrogeno e se ne aggiunge uno di carbonio si ottiene l’etano:
C2H6
- Butano: C4H10
- alcheni: CnH2n, sono idrocarburi insaturi che possono essere idrogenati saturandoli con l’aggiunta di idrogeno:
- Etilene: deriva dall’etano
- 1-butene, deriva dal butano, 1 sta ad indicare che il doppio legame si trova nel gruppo più a destra.
- 2-butene:
- dieni: CnH2n-2. Se i due doppi legami sono separati da un legame semplice si tratta di un doppio legame coniugato, se sono separati da più di un legame semplice si tratta di doppi legami isolati.
- 1,3-butadiene:
- Isoprene, uno dei “mattoni” preferiti dalla natura:
- aromatici
L’olio d’oliva è composto essenzialmente da grassi insaturi (con molti doppi legami) e si trova liquido a temperatura ambiente. Se lo si idrogena (satura) si ottiene un grasso solido (molto più dannoso per la salute), la margarina.
Composti ciclici: sono quelli che formano una struttura chiusa:
Il loro nome si forma aggiungendo il prefisso ciclo al nome del composto a catena aperta corrispondente:
ciclopropano
ciclobutano
ciclopentene
ciclopentano
Gli stessi composti possono essere rappresentati omettendo il gruppo CH2:
ciclopropano
Tra gli idrocarburi ciclici si trovano gli aromatici:
benzene
Il doppio legame può anche “ruotare” però è sempre alternato ad un legame singolo.
I composti elettrociclici contengono anche altri elementi (N, O, S)
Le molecole complesse sono composte da “mattoni” come quelli finora descritti.
I gruppi funzionali
I gruppi funzionali sono gruppi chimici che caratterizzano determinate sostanze:
Alcoli: sono caratterizzati dal gruppo funzionale ossidrile –OH. La loro formula generale è R-OH dove R rappresenta un gruppo alchilico o arilico. Vengono classificati in:
- Primari quando il gruppo ossidrile è legato ad un atomo di carbonio che non è legato a nessun altro o è legato ad un solo atomo di carbonio:
CH3-OH (metanolo)
CH3-CH2-OH (alcol etilico)
- Secondari se l’atomo di carbonio cui è legato l’ossidrile è legato a 2 altri atomi di carbonio, terziari se è legato a 3 atomi di carbonio:
isopropanolo
Aldeidi e chetoni:
Sono caratterizzati da un gruppo carbonilico, CO
- Aldeidi:
esempio: l’acetaldeide prodotta per ossidazione dell’etanolo. L’etanolo in sé non è tossico ma l’acetaldeide si ed è quella che da i postumi della sbronza:
CH3CHO.
Il glucosio ha la “funzione aldeica” nella molecola
- I chetoni hanno due gruppi alchilici legati al carbonio carbonilico:
esempio: l’acetone CH3COCH3
Il fruttosio ha la “funzione chetosa”
Sia glucosio che fruttosio però hanno formula bruta C6H12O6. Cambia solo la disposizione.
Acidi carbossilici:
possiedono nella molecola il gruppo funzionale carbossilico R-COOH
In acqua dissociano: R-COOH -> R-COO—H+, dallo ione H+ la loro natura acida.
I loro nomi hanno solitamente desinenza ico e gli ioni corrispondenti ato: acido acetico e acetato e acido lattico e lattato
Anidridi acide
Si formano in seguito alla reazione di due molecole di un acido (organico, inorganico o organico + inorganico – misto). con perdita di una molecola d’acqua. Sono composti altamente instabili che liberano grandi quantità di energia se idrolizzati:
anidride inorganica (tutti e due i reagenti non contengono carbonio).
Esteri
Sono composti che si formano per reazione reversibile tra 1 acido carbossilico ed 1 alcol con eliminazione di una molecola d’acqua:
La reazione da sinistra verso destra si chiama esterificazione, quella da destra verso sinistra idrolisi.
Ammine e ammidi:
Le ammine R-NH2 sono derivati organici dell’ammoniaca (NH3) e possono essere primarie, secondarie o terziarie secondo quanti R sono legati all’azoto:
- Primaria: R-NH2
- Secondaria:
- Terziaria:
Gli ioni che possiedono 4 gruppi R legati all’azoto sono ioni positivi che si chiamano ioni ammonici quaternari:
Le ammidi contengono nella molecola il gruppo funzionale:
e derivano dall’eliminazione di una molecola d’acqua tra un acido carbossilico e un’ammina:
RCOOH + R’NH2 -> RCONHR’ + H2O
La vitamina PP (pellagra preventing) è l’ammide dell’acido nicotinico.
Quesiti (corretti con l’aiuto di Alberto Rambaudi):
- Quale dei seguenti composti non può essere un acido?
- H2O
- CuSO4 (risposta giusta, è l’unico che non ha atomi di idrogeno da cedere).
- NaHCO3
- CH3COOH
- Qual è l’acido nella seguente reazione HNO3 + NH4OH -> NH4NO3 + H2O?
Risposta: HNO3 perché cede l’atomo di H.
- Nella reazione tra acido iodidrico e acqua qual è lo ione idronio?
HI + H2O -> H3O+ + I-
Risposta: H3O+ è lo ione idronio, I- è lo ione ioduro.
- Quale delle seguenti indica una soluzione neutra?
- pH = 7 (si)
- [H+] = 107 (è un valore enorme)
- [H+] > 107 (è un valore enorme)
- [OH+] = 10-7 (l’esponente * -1 = 7)
- Una soluzione è acida se:
- pH = 7 (no, è neutra)
- pH < 7 (si)
- [H+] > 10-7 (si, valori dell’esponente minori rappresentano numeri più grandi, gli esponenti * -1 danno valori inferiori a 7)
- [H+] < 10-7 (no, valori dell’esponente maggiori rappresentano numeri più piccoli, gli esponenti * -1 danno valori superiori a 7).
- Quanti legami idrogeno può formare la molecola d’acqua? 4
- Quanti legami idrogeno può ricevere una molecola d’acqua? 2
- Quanti legami idrogeno può donare una molecola d’acqua? 2
- L’acido è una sostanza che:
- Cede OH- (no)
- Acquista H+ (no)
- Cede H+ (si)
- Alcanilizza un ambiente (no)
- Il pH del sangue è
- Leggermente acido (no)
- Leggermente basico (si)
- Rigorosamente neutro (no)
- Il valore del pH del sangue è:
- 6,8 ± 0,2 (no)
- 7,4 ± 0,03 (si)
- 7,0 ± 0,03 (no)
- L’anidride carbonica è
- Un acido (no)
- Un gas (si)
- CO32- (no)
- O=C=O (si)
- La seguente relazione è quella corretta
- H2O + CO2 = H2O2 + CO (no)
- H2O + CO2 = H2O + CO2 (no)
- H2O + CO2 = H2CO3 (si)
- H2O + CO2 = H2CO + O2 (no)
- L’anidride carbonica disciolta nel plasma abbassa il pH? (Si)
- L’anidride carbonica costituisce il più importante sistema tampone del sangue? (Si)
- L’acido carbonico dissocia nel seguente modo:
- H2CO3 = H+ + HCO3- (si)
- H2CO3 = H2+ + HCO3-2 (no)
- Il tampone fosfato è rappresentato dal seguente sistema:
- H3PO4 = H+ + H2PO4- (no)
- H2PO4- = H+ + HPO42- (si)
- H3PO4 = 3H+ + PO43- (no)
- Il propano ha formula bruta: C3H8
- Il cicloesano ha formula bruta: C6H12
- Il benzene ha formula bruta: C6H6
- ome si chiamano i seguenti gruppi funzionali?:
- aldeide
- chetone
- R-CH2-OH alcolico 1’
- alcolico 2’
- Qual’è il nome dei seguenti composti?
- CH3CHO acetaldeide
- (CH3)2CO acetone
- CH3CH2OH etanolo
- Qual’è la formula bruta
- del fruttosio: C6H12O6
- del glucosio: C6H12O6
- Come si chiamano i seguenti gruppi funzionali:
- RCOOH: acido carbossilico
- RCOOOCR’: anidride organica
- RCOOPO3H2: anidride mista
- RCOOR’: estere
- Quanti gruppi OH contiene il glicerolo? 3
3.06 Spiegare che differenza c’è tra un legame carbamidico e un legame peptidico: il legame carbamidico avviene tra un gruppo amminico ed un carbossilico per eliminazione di una molecola d’acqua. Quello peptidico tra un gruppo alfa-amminico ed un alfa-carbossilico per eliminazione di una molecola d’acqua.
- Da un punto di vista chimico la vitamina PP è? Un ammide dell’acido nicotinico.
4.01 Definire la caloria: la quantità di energia necessaria ad aumentare la temperatura di 1°C di 1g di acqua tra 14,5 e 15,5°C.
4.02 Definire il legame covalente: legame forte tra atomi con la stessa elettronegatività.
4.03 Definire il legame ionico: legame tra atomi con carica elettrica opposta.
4.04 Definire il legame idrogeno: legame con un atomo di idrogeno interposto tra due atomi elettronegativi.
4.05 Definire il legame idrofobico: interazione che avviene tra gruppi a carattere idrofobico.
4.06 Quali gruppi partecipano alla formazione del legame peptidico: alfa-carbossilico e alfa-amminico.
4.07 Definire la struttura primaria delle proteine: semplice sequenza degli amminoacidi a partire dall’amminoterminale al carbossiterminale.
4.08 Descrivere la struttura dell’insulina: due catene polipeptidiche di 21+30 aminoacidi legate tra loro tra due ponti “disolfuro”.
4.09 Cosa si intende per sostituzione conservativa e non conservativa: conservativa è quanto un aminoacido viene sostituito con un altro avente analoghe caratteristiche chimiche. Non conservativa quando l’aminoacido che sostituisce ha caratteristiche chimiche diverse.
4.10 L’alfa-elica è stabilizzata da: legami a idrogeno all’interno della stessa catena.
4.11 Il foglietto beta è stabilizzato da: legami a idrogeno tra catene diverse.
4.12 L’aminoacido più abbondante nel collagene è: la glicina.
4.13 Gli aminoacidi caratteristici della struttura del collagene sono: glicina, prolina, idrossiprolina.
4.14 La struttura della mioglobina è la seguente: 153 aminoacidi organizzati in 8 alfaeliche numerate da A ad H.
4.15 La mioglobina ha struttura? Terizaria.
4.16 L’emoglobina ha struttura? Quaternaria.
4.17 La struttura dell’emoglobina è la seguente: 4 subunità (2 alfa e 2 beta) 141 aminoacidi alfa e 146 aminoacidi beta.
5.01 Le emoproteine sono costituite da: globina (parte proteica) e eme (parte prostetica).
5.02 Che cosa si intende per globina? La parte proteica delle emoproteine.
5.03 Cosa si intende per EME? La parte prostetica delle emoproteine.
5.04 I tratti ad alfa-elica della Mb sono: 8.
5.05 La Mb ha struttura secondaria: falso
5.06 La Mb ha struttura terziaria: vero
5.07 I tratti ad alfa-elica della catena alfa dell’Hb sono: 7
5.08 I tratti ad alfa-elica della catena beta dell’Hb sono: 8
5.09 Il numero di coordinazione del Fe è: 6
5.10 Il ferro nell’eme contrae legami di coordinazione con: i quattro azoti dell’anello porfirinico e i due azoti dell’istidina prossimale e distale.
5.11 Come l’eme è legato alla globina? Con il legame dell’istidina prossimale F8 e distale E7.
5.12 Lo stato di ossidazione del ferro nell’eme è: 2+
5.13 Quando l’ossigeno si lega all’eme lo stato di ossidazione del Fe passa da 2+ a 3+ FALSO.
5.14 Il legame dell’ossigeno con l’emoglobina avviene attraverso: il 6° legame di coordinazione.
5.15 La pressione dell’ossigeno alveolare è: 100 mm Hg
5.16 La p50 dell’ossigeno nel muscolo a riposo è 40mm Hg
6.01 Perché i carboidrati sono impropriamente detti idrati di carbonio? Perché presentano atomi di carbonio legati a atomi di idrogeno e a gruppi ossidrilici.
6.02 Quanti gruppi alcolici secondari sono presenti nella molecola di glucosio? 4
6.03 Quanti gruppi alcolici secondari sono presenti nella molecola del ribosio? 3
6.04 Quanti gruppi alcolici secondari sono presenti nella molecola del fruttosio? 3
6.05 Il glucosio è? Un aldoesoso.
6.06 Il fruttosio è? Un chetoesoso.
6.07 Il ribosio è? Un aldopentoso.
6.08 L’anello emiacetalico tra il C2 e il C5 appartiene a? Fruttosio.
6.09 L’anello emiacetalico tra il C1 e il C5 appartiene a? Glucosio.
6.10 Nel ribosio l’anello emiacetalico si forma tra il C1 e il C4: vero.
6.11 Il galattosio è l’epimero in C4 del glucosio.
6.12 Il saccarosio è un? Disaccaride.
6.13 Nel saccarosio, fruttosio e glucosio sono uniti da un legame? Alfa-Glc (1<->2) beta-Fru.
6.14 Il lattosio è costituito da: BetaGal (1<->4) AlfaGlc
6.15 Il maltosio è: AlfaGlc (1<->4) alfaGlc
6.16 Il glicogeno ha struttura: ramificata
7.01 Una reazione si dice esergonica quando: (c)l’energia libera dei prodotti è inferiore all’energia libera dei reagenti.
7.02 Una reazione si dice endergonica quando: (d)l’energia libera dei reagenti è inferiore all’energia libera dei prodotti.
7.03 Si definisce energia di attivazione: (a)la differenza tra l’energia dello stato di attivazione e l’energia dei reagenti.
7.04 La velocità di una reazione dipende: dalla temperatura del sistema, dalla concentrazione dei reagenti e dalla presenza di un catalizzatore.
7.05 Gli enzimi sono catalizzatori inorganici: (b) falso.
7.06 Definire la specificità stereochimica: quando il substrato esiste in più forme isomere e l’enzima ne predilige una.
7.07 Definire la specificità di legame: Quando l’enzima si lega ad un determinato legame a prescindere dalla natura dei gruppi chimici che lo costituiscono.
7.08 Definire la specificità di gruppo: Quando l’enzima si lega solo in presenza di un determinato legame e gruppo chimico che lo costituisce.
7.09 Definire la specificità assoluta: Quando un enzima riconosce sia il legame che i due gruppi chimici che unisce.
7.10 Definire il sito attivo di un enzima: La parte che entra in contatto col substrato.
7.11 L’interazione enzima substrato si può spiegare attraverso due modelli. Quali?
- Chiave - serratura.
- Adattamento indotto.
7.12 Data la seguente sequenza aminoacida: ---X-ALA-SER-ARG-GLY-TRP-GLU-PHE-X---.
- Indicare il sito di taglio della tripsina: ARG-GLY (d)
- Della chimotripsina: -GLU-PHE- (f-h)
7.13 Il tripsinogeno è attivato a tripsina ad opera di un enzima detto? Enterochinasi.
7.14 La funzione della vitamina K è quella di? Favorire la trasformazione dell’ac.γ-carbossi-Glu.
7.15 Quando l’inibitore si lega al sito attivo dell’enzima si ha un’inibizione di tipo: competitivo.
7.16 Quando l’inibitore si lega all’enzima in un sito diverso dal sito attivo, si ha un’inibizione di tipo: non competitivo.
8.01 Gli enzimi si dividono in sei classi principali, quali?
- ossidoriduttasi
- transferasi
- idrolasi
- liasi
- isomerasi
- ligasi
8.02 Con il termine ossidazione si intendono i seguenti tipi di reazioni:
- Addizione ossigeno.
- Sotrazione idrogeno.
- Sottrazione di elettroni.
8.03 Le deidrogenasi si dividono in due sottoclassi, quali? Piridiniche e flaviniche.
8.04 Le ossidoriduttasi si dividono in cinque sottoclassi, quali?
- Deidrogenasi;
- Ossidasi;
- Perossidasi;
- Ossigenasi;
- Trasporto elettrone.
8.05 Cosa si intende per NAD? Nicotinamide adenin dinucleotide.
8.06 Cosa si intende per FAD? Flavin adenin dinucleotide.
8.07 Nella reazione di riduzione del NAD+ il substrato: (c) perde 2 elettroni + 2 protoni.
8.08 Quali sono i componenti che costituiscono la struttura del NAD? Nicotinamide, 2 ribosi, 2 fosfat1, adenina.
8.09 Nella reazione di riduzione del FAD il substrato: (c) perde 2 elettroni + 2 protoni.
8.10 Quali sono i componenti che costituiscono la struttura del FAD? flavina, adenina 2 ribosi, 2 fosfati.
8.11 Qual’è la vitamina che costituisce il coenzima flavinico? B2.
8.12 Qual’è la vitamina che costituisce il coenzima piridinico? PP.
8.13 Qual’è il ruolo biochimico della niacina (PP)? E’ la parte vitaminica del cofattore NAD.
8.14 Qual’è il ruolo biochimico della riboflavina (vit. B2)? E’ la parte vitaminica del cofattore FAD.
9.01 Qual’è il cofattore della transaminasi? PLP
9.02 A quale classe enzimatica appartengono le transaminasi? Transferasi
9.03 Qual’è la vitamina che costituisce il PLP? B6
9.04 I prodotti della reazione di transaminazione tra Glu e ac. ossalacetico catalizzata dalla GOT sono:
- b. alfa-ketoglutarato
- c. Asp
9.05 I prodotti della reazione di transaminazione tra Glu e ac. piruvico catalizzata dalla GPT sono:
- a. Ala
- b. alfa-ketoglutarato
9.06 Qual’è i donatore di metili nelle reazioni catalizzate dalle transmetilasi? SAM
9.07 Qual’è la funzione della fosfocreatina? Ricostituire l’ATP dall’ADP.
9.08 La reazione ADP + ADP <-> AMP + ATP è catalizzata da quale enzima? Miochinasi
9.09 Qual’è la funzione dell’esochinasi? fosforilare il glucosio in posizione 6 nel muscolo
9.10 Qual’è la funzione della glucochinasi? fosforilare il glucosio in posizione 6 nel fegato.
9.11 Il legame dell’adrenalina sul proprio recettore di una cellula muscolare induce:
- b. attivazione della glicogeno fosforilasi.
9.12 Il prodotto della degradazione fosforolitica del Gn ad opera della glicogenofosforilasi è:
- b. G-1-P
9.13 Nella sequenza polipeptidica -ASP-TYR-AAx-LYS-AAx- indicare con una freccia il sito di taglio della pepsina. Tra ASP e TYR.
9.14 Nella sequenza polipeptidica -ASP-TYR-AAx-LYS-AAx- indicare con una freccia il sito di taglio della chimotripsina. Tra TYR e AAx.
9.15 Nella sequenza polipeptidica -ASP-TYR-AAx-LYS-AAx- indicare con una freccia il sito di taglio della tripsina. Tra LYS e AAx.
9.16 Le lipasi pancreatiche idrolizzano i trigliceridi in glicerolo + acidi grassi.
9.17 Scrivere nella corretta sequenza gli enzimi che partecipano alla degradazione del glicogeno in glucosio ematico.
- Gn fosforilasi.
- Fosfoglicomutasi.
- G-6-Pasi.
9.18 Il cofattore della reazione di decarbossilazione dell’acido piruvico è il DPT derivante della vitamina B1.
9.19 Completare la seguente reazione catalizzata dalla miochinasi:
- ADP + ADP <-> ATP + AMP.
9.20 Completare la seguente reazione catalizzata dalla esochinasi:
- ATP + glucosio -> ADP + G6P.
9.21 Completare la seguente reazione catalizzata dalla glucochinasi:
- ATP + glucosio -> ADP + G6P.
9.22 Completare la reazione catalizzata dalla glicogeno fosforilasi:
- Gn + P -> Gn-1 + G1P.
9.23 Completare la reazione catalizzata dalla glicogeno fosforilasi:
- Gn + P -> Gn-1 + G1P.
9.24 Il G-6-P può derivare da:
- G + P: falso
- G + ATP: vero
- G-1-P: vero
10.01 Quali reazioni catalizzano gli enzimi appartenenti alla classe delle LIASI? Catalizzano il distacco o legame di un frammento chimico su un substrato.
10.02 Qual’è il cofattore delle alfa-chetoacido decarbossilasi? DPT
10.03 Qual’è il cofattore delle aminoacido decarbossilasi? PLP
10.04 Qual’è il ruolo biochimico della tiamina? Costituente essenziale del cofattore DPT.
10.05 Definire la classe di enzimi ISOMERASI: enzimi che trasformano il substrato in un isomero.
10.06 Il DHAP è trasformato reversibilmente in Gal-3 dall’enzima: triosofosfatoisomerasi.
10.07 Il G-6-P è trasformato reversibilmente in F-6-P dall’enzima: fosfoesosoisomerasi.
10.08 Definire la classe di enzimi LIGASI: enzimi che catalizzano il legame tra due composti.
10.09 Completare la seguente reazione catalizzata dall’acil-CoA sintetasi: R-COOH + CoA-SH + ATP -> RCO-SCoA + AMP + PP
10.10 Completare la seguente reazione catalizzata dalla piruvico carbossilasi: CH3-CO-COOH + CO2 + ATP -> acido salacetico + ADP + P.
10.11 La vitamina H è il cofattore di quali enzimi? Carbossilasi.
10.12 L’albume d’uovo contiene una proteina in grado di impedire l’assorbimento intestinale della biotina, come si chiama? Avidina.
10.13 L’acido pantotenico è la vitamina che costituisce un importante coenzima, quale? CoA.
10.14 Qual’è la funzione del CoA? Trasportare gli acili.
10.15 Il beta-carotene è il precursore di quale vitamina? A
10.16 In quante forme è presente in natura la vitamina A? 3 Come si chiamano? Retinolo, retinale, acido retinoico.
10.17 La conversione dell’11-cis-retinale in tutto-trans, in quale meccanismo biochimico si ritrova? Visione crepuscolare.
10.18 Che differenza c’è tra opsina e rodopsina? La rodopsina è la proteina coniugata costituita da opsina + retinale-11-cis.
10.19 Qual’è la reazione chiave che innesca l’impulso nervoso nei fotorecettori dei bastoncelli presenti nella retina? La conversione dell’11-cis-retinale in retinale tutto-trans.
10.20 Da dove deriva la vitamina D3 o colecalciferolo? Dal 7-deidrocoloesterolo attraverso l’apertura dell’anello B grazie ai raggi UV.
10.21 Che differenza c’è tra il 7-deidrocolesterolo e il colecalciferolo? L’apertura dell’anello B attraverso l’irraggiamento UV.
10.22 Qual’è la forma biologicamente attiva della vitamina D? 1-25-(OH2)D3
10.23 Qual’è la funzione della vitamina D? regolare l’omeostasi del calcio oppure favorire l’assorbimento intestinale di Ca.
10.24 Descrivere il processo biochimico che porta alla formazione dell’1-25-(OH2)D3. Il 7-deidrocolesterolo irraggiato da raggi UV diventa vitamina D3 che viene idrossilata nel fegato in posizione 25 e nel rene in posizione 1 a formare l’1-25-(OH)2-D3.
10.25 Che si intende per omeostasi del Ca++? Il mantenimento della concentrazione ematica.
10.26 Quali fattori e in che modo partecipano all’omeostasi del calcio? 1-25-(OH2)D3, PTH, calcitonina.
10.27 Qual’è la funzione del paratormone? Promuovere l’assorbimento intestinale del Ca++ ed il rilascio di esso dalle ossa.
10.28 Qual’è la funzione della calcitonina? Calciofissatrice.
11.01 Com’è costituito l’ATP? Adenina + ribosio + 3 fosfati.
11.02 Quanti e quali sono i legami ricchi di energia presenti nell’ATP? 2 tra fosfato alfa e beta e tra fosfato beta e gamma.
11.03 Qual è la funzione dell’ATP? Fornire energia ai processi vitali.
11.04 Quali sono rispettivamente i prodotti della glicolisi:
- In un muscolo che lavora intensamente? Lattato.
- In un muscolo a riposo? Acido piruvico.
- Nell’eritrocita? Lattato.
11.05 Quante molecole di ATP vengono consumate durante la glicolisi? 2
11.06 Quante molecole di ATP si producono al netto durante la glicolisi? 2
11.07 Quanto ATP produce la glicolisi eritrocitaria? 2
11.08 Qual è il prodotto della glicolisi eritrocitaria? Lattato.
11.09 Qual è il coenzima della gliceraldeide-3-fosfato deidrogenasi? NADH
11.10 In un muscolo che lavora in condizioni di carenza di ossigeno, come viene riossidato il NADH prodotto durante la degradazione glicolitica del glucosio? Attraverso la fermentazione lattica.
11.11 In normali condizioni fisiologiche, come viene riossidato il NADH prodotto durante la degradazione glicolitica del glucosio? Attraverso la catena del trasporto mitocondriale.
11.12 Cosa si intende per fermentazione lattica? La riossidazione del NADH prodotta dalla GAP-DH con riduzione del piruvato al lattato.
11.13 Cosa si intende per fermentazione alcolica? La riossidazione del NADH prodotta dalla GAP-DH con riduzione del piruvato ad etanolo.
11.14 Quali enzimi partecipano alla digestione dell’amido? alfa amilasi e alfa(1->6) glicosidasi
11.15 Cosa si produce per azione dell’alfa amilasi sull’amiloso? Il maltosio.
11.16 Cosa si produce per azione dell’alfa amilasi e della alfa(1->6) glicosidasi sull’amilopectina? Maltosio + glucosio.
11.17 La degradazione del glicogeno epatico produce: glucosio ematico.
11.18 La degradazione del glicogeno muscolare produce: G1P per il metabolismo muscolare.
11.19 Qual è il contributo delle diverse sorgenti di energia durante un’attività muscolare di media intensità:
- Nei primi secondi: ATP
- Entro i primi 30 secondi: PC
- Entro il primo minuto: metabolismo anaerobico.
- Per tempi più lunghi: metabolismo aerobico.
12.01 Quante molecole di CO2 si producono durante il ciclo di Krebs per ossidazione di una molecola di acetil-CoA? 2.
12.02 Quante molecole di NADH e quante di FADH2 si producono durante il ciclo dell’acido citrico? 3 + 1.
12.03 Nel ciclo dell’acido citrico quante molecole di ATP si producono durante l’ossidazione di una molecola di acetil-CoA? 1 a livello del substrato e 11 dalla riossidazione di NADH e FADH2.
12.04 Quali sono le fonti metaboliche dell’acetil-CoA per il ciclo degli acidi tricarbossilici? Acidi grassi, carboidrati, amminoacidi.
12.05 In quale compartimento cellulare sono contenuti gli enzimi del ciclo di Krebs? All’interno del mitocondrio.
12.06 Qual è l’origine dell’ossalacetato per il ciclo dell’acido citrico? L’acido piruvico.
12.07 Come vengono riossidati il NADH ed il FADH2 prodotti durante il ciclo di Krebs? Attraverso la catena di respirazione.
12.08 Dov’è localizzato il sistema della catena respiratoria? Nella membrana interna del mitocondrio.
12.09 Qual è la funzione della catena respiratoria? Riossidare le catene piridiniche e flaviniche.
12.10 Quante molecole di ATP si producono nella fosforilazione ossidativa per ossidazione del NADH e quante per ossidazione del FADH2? 3 dal NADH e 2 dal FADH2
12.11 Cosa si intende per fosforilazione ossidativa? ADP + P -> ATP
12.12 Qual’è l’accettore terminale degli elettroni nella catena respiratoria? L’ossigeno.
12.13 Qual’è il prodotto di riduzione dell’O2 nella catena respiratoria? Acqua
12.14 Cosa si intende per beta-ossidazione? Processo di riossidazione del beta carbonio degli acidi grassi.
12.15 Quali sono i prodotti della beta-ossidazione dell’acido palmitico? 8 AcetilCoA + 7 NADH + 7 FADH2.
12.16 Quanto ATP si produce per ossidazione dell’acido palmitico? 129
12.17 Qual’è il ruolo della carnitina? Trasportare acili dal citosol alla matrice del mitocondrio.
12.18 Come viene trasportato l’acile nel motocondrio? Attraverso la carnitina.
12.19 Cosa si intende per quoziente respiratorio? Il rapporto tra anidride carbonica prodotta e ossigeno consumato nel metabolismo.
12.20 Qual’è il Q.R. del metabolismo ossidativo dei carboidrati e dei grassi?
- Q.R. carboidrati= 1
- Q.R. grassi= 0,7
13.01 Quali sono i principali tessuti glucogenetici? Fegato, corticale renale.
13.02 Cosa si intende per glucogenesi? Sintesi del glucosio da materiale di natura non strettamente glicinica.
13.03 Quante molecole di ATP vengono consumate nella glucogenesi? 6.
13.04 Descrivere brevemente il Ciclo di Cori: l’acido lattico prodotta dal muscolo viene trasportata nel fegato dove viene usata per produrre glucosio che torna nei muscoli per ripristinare le riserve di glciogeno.
13.05 Descrivere brevemente il ciclo glucosio-alanina: l’ammoniaca e lo scheletro carbonoso del piruvato passano dal muscolo al fegato attraverso l’alanina. L’amina viene secreta mentre il piruvato usato per produrre glucosio che ritorna ai muscoli.
13.06 In quali condizioni metaboliche la chetogenesi è particolarmente attiva? Nel digiuno prolungato e nel diabete.
13.07 Qual’è il substrato per la sintesi degli acidi grassi? Acetil-CoA
13.08 Attraverso quale composto l’acetile passa dalla matrice mitocondriale al citosol per dare inizio alla sintesi degli acidi grassi? Il citrato.
13.09 Qual’è il coenzima delle riduttasi impegnate nella sintesi degli acidi grassi? NADPH.
13.10 Qual’è l’origine metabolica dell’acetil-CoA per la sintesi degli acidi grassi? Carboidrati e proteine.
13.11 Qual’è l’origine del glucosio ematico dopo un digiuno prolungato? La glucogenesi.
13.12 Qual’è l’origine del glucosio ematico nelle prime fasi del digiuno? La degradazione gel glicogeno epatico.
14.01 Qual’è il normale destino metabolico dello scheletro carbonioso degli amminoacidi dopo la rimozione dell’azoto? Il ciclo di Krebs.
14.02 Contrassegnare tra i seguenti amminoacidi quelli glucogenici: (a) aspartato e (c) istidina.
14.03 Contrassegnare tra i seguenti amminoacidi quelli chetogenici: (b) leucina e (d) lisina.
14.04 In quale organo avviene principalmente l’ureogenesi? Il fegato.
14.05 Quanti atomi d’azoto contiene la molecola dell’urea? 2.
14.06 Quante molecole di ATP si consumano per la sintesi dell’urea? 4.
14.07 La tappa biosintetica fondamentale comune a tutti i processi di sintesi di neurotrasmettitori derivati da amminoacidi è: carbossilazione PLP dipendente.
14.08 Da quale amminoacido deriva l’adrenalina? La tirosina.
14.09 Assegnando una numerazione ai seguenti composti, metterli nel corretto ordine metabolico: (5) Adrenalina (2) Dopa (1) Tirosina (3) Dopamina (4) Noradrenalina
14.10 Attraverso quale reazione e da quale amminoacido deriva il GABA? La carbossilazione PLP dipendente dell’acido glutamminico: glutammato.
14.11 Attraverso quale reazione e da quale amminoacido deriva l’istamina? La carbossilazione PLP dipendente dell’istidina.
14.12 Da quale amminoacido deriva la serotonina? Il triptofano.
14.13 Qual’è il contenuto calorico medio in kcal/g di
(a) carboidrati: 4
(b) lipidi: 9
(c) proteine: 4
15.01 Qual’è la differenza tra un nucleoside ed un nucleotide? Il nucleoTide è un nucleoSide fosforilato.
15.02 Com’è costituito l’AMP? Da adenina + ribosio + fosfato.
15.03 C’è differenza c’è tra AMP e AMPc? Il cAMP, il fosfato esterifica in 5’ e in 3’ mentre l’AMP fosforila solo in posizione 5’.
15.04 Quali sono le basi puriniche? Adenina, guanina.
15.05 Quali sono le basi pirimidiniche? Citosina, timina, uracile.
15.06 Qual’è lo zucchero presente nella struttura del DNA? Desossiribosio.
15.07 Qual’è lo zucchero presente nella struttura del RNA? Ribosio.
15.08 Scrivere la sequenza complementare alla seguente -A-T-G-C-C: -T-A-C-G-G-
15.09 Scrivere la sequenza complementare alla seguente -A-U-G-C-C: -U-A-C-G-G-
15.10 Quanti legami di idrogeno si possono formare tra A e T? 2
15.11 Quanti legami di idrogeno si possono formare tra G e C? 3
15.12 Da cosa è stabilizzata la struttura a doppia elica del DNA? Legame idrogeno.
15.13 Nel processo di duplicazione del DNA vengono usati: (b) nucleosidi trifosfato.
15.14 Cosa si intende per tripletta? La sequenza di 3 basi.
15.15 Cos’è il codice genetico? La corrispondenza tra tripletta e amminoacido.
15.16 Qual’è la funzione del mRNA? Portare dal nucleo al ribosoma l’informazione per la sintesi proteica.
15.17 Qual’è la funzione del tRNA? Portare l’amminoacido al ribosoma.
15.18 Cosa si intende per traduzione? Tradurre la sequenza del mRNA in proteina.
Domande a svolgimento libero:
- Descrivere il trasporto dell’ossigeno e l’effetto Bohr.
- Descrivere il trasporto dell’anidride carbonica.
- Descrivere il meccanismo della visione.
- Descrivere il meccanismo che regola l’omoeostasi del calcio.
- Descrivere il processo di degradazione del glicogeno epatico e muscolare.
- Descrivere l’utilizzazione del glucosio, aminoacidi e lipidi nei vari tessuti nella fase di buona alimentazione.
- Descrivere le interrelazioni metaboliche fra i principali tessuti dell’organismo durante la fase iniziale di digiuno.
- Descrivere le interrelazioni metaboliche fra i principali tessuti dell’organismo durante la fase di digiuno.
- Descrivere le interrelazioni metaboliche fra i principali tessuti dell’organismo durante la fase iniziale di rialimentazione.
- Descrivere il processo fondamentale del trasferimento dell’informazione nelle cellule.
Autore: Appunti di Luca Asberto
Fonte: http://www.asberto.net/Anno%203/Biochimica.doc
Biochimica
Visita la nostra pagina principale
Biochimica
Termini d' uso e privacy