Chimica appunti parte 3
Chimica appunti parte 3
Questo sito utilizza cookie, anche di terze parti. Se vuoi saperne di più leggi la nostra Cookie Policy. Scorrendo questa pagina o cliccando qualunque suo elemento acconsenti all’uso dei cookie.I testi seguenti sono di proprietà dei rispettivi autori che ringraziamo per l'opportunità che ci danno di far conoscere gratuitamente a studenti , docenti e agli utenti del web i loro testi per sole finalità illustrative didattiche e scientifiche.
L'effetto tunnel
Un'esemplificazione concreta delle bizzarrie quantistiche è data dal cosiddetto effetto tunnel, che comporta la materializzazione di particelle in regioni ad esse inaccessibili secondo le leggi della fisica classica.
Immaginiamo una sfera posta all'interno di un recipiente. Se sulla sfera non agisce nessuna forza essa non potrà assolutamente uscire.
Nella teoria quantistica però la particella viene descritta da un'onda di probabilità interna al recipiente, onda il cui quadrato esprime la probabilità di trovare la particella.
Si può dimostrare che se nel recipiente si trova ad esempio un elettrone, l'onda di probabilità ad esso associata si prolunga, sia pur di poco, all'esterno delle pareti del recipiente. Ne segue che l'elettrone possiede una probabilità minima, ma finita, di manifestarsi all'esterno delle pareti del recipiente. Se noi effettuassimo una serie di osservazioni troveremmo perciò l'elettrone quasi sempre all'interno del recipiente, ma in alcuni rari casi anche fuori.
L'effetto tunnel viene utilizzato ormai normalmente nell'ingegneria elettronica per amplificare i segnali elettronici.
L'attraversamento quantistico di una barriera di potenziale contribuisce altresì a giustificare il fenomeno della radioattività, dove il nucleo emette spontaneamente particelle che per la fisica classica dovrebbe trattenere.
L'effetto tunnel è stato invocato anche in astrofisica da S. Hawking per sostenere la sua teoria dell'evaporazione dei buchi neri.
Il gatto di Schrödinger ed il principio di sovrapposizione degli stati
In Meccanica Quantistica le grandezze fisiche che caratterizzano un sistema e che possono essere misurate (posizione, velocità, energia, momento magnetico, eccetera) sono chiamate osservabili.
I possibili valori che può assumere un’osservabile definiscono i potenziali stati in cui il sistema può presentarsi, detti autostati. Soltanto all'atto della misurazione fisica si può ottenere un valore reale per gli osservabili. Fintantoché non si esegue la misura il sistema quantistico rimane in uno stato che è "oggettivamente indefinito", sebbene sia matematicamente definito e costituito dalla sovrapposizione di tutti gli stati possibili. Lo stato del sistema prima della misura descrive solo una "potenzialità" ovvero contiene l'informazione relativa ad una "rosa" di valori possibili (stati di sovrapposizione), ciascuno con la sua probabilità di divenire reale ed oggettivo all'atto della misura.
In altre parole, il sistema sta potenzialmente in tutti gli stati contemporaneamente. Il suo stato diventerà "puro", unico, solo dopo e come conseguenza di una misura o di un'interazione con un altro sistema.
Nel linguaggio della meccanica quantistica, si dice che all'atto della misura dell'osservabile lo stato collassa in uno dei tanti possibili autostati ammessi da quell'osservabile. Il passaggio di un sistema fisico dal suo stato indeterminato di sovrapposizione ad un particolare autostato si definisce collasso o riduzione. All'atto della misurazione l’incertezza probabilistica viene ridotta o collassa nella certezza di un numero ben determinato. L'osservazione del fenomeno diviene quindi parte fondamentale della medesima realtà che si vuol misurare.
Proviamo a vedere un semplice esempio. Consideriamo un elettrone che si trova in un certo sistema fisico e cerchiamo di misurare la sua energia in un dato istante. Prima della misura, esso non avrà un'energia definita, ma si troverà in uno stato potenziale che contiene (ad esempio):
- l'autostato di energia 850 eV, con probabilità del 20%;
- l'autostato di energia 860 eV, con probabilità del 35%;
- l'autostato di energia 870 eV, con probabilità del 45%.
All'atto della misura del valore dell'energia, la natura dovrà "scegliere" uno dei tre possibili "autostati" dell'energia, ciascuno dei quali ha il suo valore (chiamato "autovalore"): 850 o 860 o 870 eV. Essi sono valori "quantizzati", ovvero discreti o discontinui (in parole povere non sono possibili valori intermedi, come 865 eV). Pertanto lo stato iniziale è oggettivamente "indefinito" rispetto all'osservabile energia, poiché è una combinazione (o sovrapposizione) di tre autostati diversi, ed all'atto della misurazione dovrà "collassare" in uno dei tre possibili "autostati", che danno valori validi dell'energia nella realtà fisica oggettiva. Ogni volta il risultato potrà essere diverso, e ciascun "autovalore" ha la sua probabilità di uscire.
La meccanica quantistica quindi introduce due elementi nuovi ed inaspettati rispetto alla fisica classica. Uno è appunto l'influenza dell'osservatore, che costringe lo stato a diventare un autostato; l'altro è la casualità nella scelta di uno tra i diversi possibili autostati (ognuno con una propria probabilità).
Einstein non credeva alla possibilità di caratteristiche fisiche "non-oggettive", ma riteneva che i valori delle osservabili esistessero oggettivamente anche prima della misura (realismo), indipendentemente dal fatto che venissero misurati o meno. Insomma, secondo Einstein l'universo deve esistere oggettivamente, sia che noi lo osserviamo o meno. Per questo egli considerava la meccanica quantistica "incompatibile con ogni concezione ragionevole e realistica dell'universo".
Famosa resta a questo proposito la domanda che egli pose ad un allievo durante una passeggiata serale a Princeton: «Veramente è convinto che la Luna esista solo se la si guarda?»
Secondo il "realismo" di Einstein, gli stati quantistici devono esistere oggettivamente, indipendentemente da tutte le limitazioni imposte dalla teoria quantistica, che perciò secondo Einstein è incompleta e provvisoria.
Una teoria fisica e' completa qualora ogni elemento della realtà descritta abbia corrispondenza con un elemento teorico. Esisterebbero quindi, secondo Einstein, delle "variabili nascoste" che descrivono la realtà oggettiva dei sistemi quantistici, ma non sono ancora riconosciute dall'attuale teoria e che, se scoperte, renderebbero completa la teoria quantistica.
Per fare un paragone banale, immaginiamo che in una partita di carte il nostro avversario abbia in mano una certa carta. Noi deduciamo che tale carta possa essere l'asso di denari o il re di cuori, ma poiché non possiamo vederla, non sappiamo quale delle due sia realmente. Questa, secondo Einstein è la "conoscenza incompleta" che ci può dare la meccanica quantistica. Comunque, dice Einstein, la carta in questione è di fatto una delle due carte, ad esempio l'asso di denari (variabile nascosta), anche se noi non sappiamo ancora per certo se sia l'una o l'altra (indeterminazione). All'atto della misura noi possiamo finalmente constatare di quale carta si tratti, ma secondo Einstein la carta era quella già prima della misura.
Secondo la meccanica quantistica invece non è così. La carta in precedenza era in uno stato indefinito: "50% asso di denari e 50% re di cuori", e solo all'atto della misura la carta è "diventata" (ad esempio) l'asso di denari. Se si ritorna a quello stesso identico stato fisico e si rieffettua la misura, stavolta la carta potrebbe diventare un re di cuori!
Il principio quantistico di sovrapposizione degli stati e le paradossali conseguenze di una sua applicazione a livello macroscopico sono l’argomento di un esperimento mentale ideato da Erwin Schrödinger nel 1935 e noto come il paradosso del gatto di Schrödinger.
Vediamolo descritto dalle stesse parole dell’autore.
« Si rinchiuda un gatto in una scatola d’acciaio insieme con la seguente macchina infernale (che occorre proteggere dalla possibilità d’essere afferrata direttamente dal gatto). In un contatore Geiger si trova una minuscola porzione di sostanza radioattiva, in quantità così modesta che nel corso di un’ora uno dei suoi atomi possa disintegrarsi (…). Se ciò accade, allora il contatore lo segnala e aziona un relais di un martelletto che rompe una fiala contenente del cianuro. Dopo avere lasciato indisturbato questo sistema per un’ora (…) la funzione Ψ dell’intero sistema porta ad affermare che in essa il gatto vivo e il gatto morto non sono stati puri, ma miscelati con uguale peso »
Dopo un certo periodo di tempo, quindi, il gatto ha la stessa probabilità di essere morto quanto l'atomo di essere decaduto. Visto che fino al momento dell'osservazione l'atomo esiste nei due stati sovrapposti, il gatto resta sia vivo sia morto fino a quando non si apre la scatola, ossia non si compie un'osservazione.
Il paradosso sta proprio qui. Finché non si compie l'osservazione, il gatto può esser descritto come un ibrido vivo-morto, in quanto è soltanto l'osservazione diretta che, alterando i parametri di base del sistema, attribuirà al gatto (al sistema medesimo) uno stato determinato e "coerente" con la nostra consueta realtà.
Volendo seguire alla lettera le regole quantistiche, se, all’apertura della scatola d’acciaio, lo sperimentatore trova il gatto morto, è necessario ammettere che è stato l’atto di guardare ("osservare") dentro la scatola che ha ucciso il gatto ed è quindi lo sperimentatore il responsabile della sua morte.
Se lo sperimentatore decide di rimandare indefinitamente l’osservazione della scatola, il gatto resta nel suo stato schizofrenico di vita latente fino a quando non gli viene data una dimensione definitiva, in virtù della cortese, ma capricciosa curiosità di uno sperimentatore
Paradosso EPR: Entanglement e Nonlocalità
Come abbiamo già avuto modo di dire, Einstein era estremamente critico nei confronti della Meccanica Quantistica (che pur aveva contribuito a fondare). Pur riconoscendo naturalmente che la teoria funzionava perfettamente sul piano sperimentale, sosteneva che si trattava tuttavia di una teoria incompleta e provvisoria, che avrebbe dovuto essere perfezionata col tempo per eliminare alcuni aspetti inaccettabili..
Secondo Einstein una teoria che descriva la realtà fisica deve soddisfare alcune condizioni, riassumibili attraverso i principi di "realismo", "località" e "completezza".
Il realismo è l'assunzione realistica che tutti gli oggetti debbano oggettivamente possedere dei valori preesistenti per ogni possibile misurazione prima che queste misurazioni vengano effettuate. La realtà oggettiva esiste a prescindere dall’atto di osservarla e misurarla.
Come conseguenza di ciò, la realtà fisica viene associata all’esistenza di opportune proprietà oggettive (elements of physical reality) e la completezzastrutturale di ogni teoria è espressa dalla corrispondenza tra queste proprietà e gli elementi teorici formali.
A questo proposito è rimasta celebre la sua frase: "Dio non gioca a dadi con il mondo". Meno famosa è la risposta di Bohr: "Non è compito degli scienziati dire a Dio come funziona il mondo, ma solo scoprirlo".
Il principio di località afferma che eventi distanti nello spazio non possono comunicare e quindi influenzarsi istantaneamente, senza alcuna mediazione. Sappiamo infatti che la massima velocità raggiungibile è quella della luce, il che comporta che il minimo ritardo possibile tra una causa ed il suo effetto è il tempo necessario affinché un segnale luminoso percorra lo spazio che li divide. Un effetto nonlocale è noto come "azione istantanea a distanza" («spooky action at a distance» o «azioni-fantasma») ed è incompatibile con il postulato alla base della relatività ristretta, che considera la velocità della luce la velocità limite alla quale può essere accelerata una massa.
Il realismo locale è la combinazione del principio di località e di realismo.
Einstein tentò più volte di scovare un punto debole all'interno della teoria quantistica. Uno dei suoi attacchi più famosi e che resistette più a lungo dando per molto tempo filo da torcere ai fisici quantistici fu il cosiddetto esperimento mentale EPR, dai nomi di coloro che lo avevano proposto nel 1935: Einstein, Rosen e Podolsky.
Gli autori intendevano dimostrare che se si accettano gli assunti della fisica quantistica veniva automaticamente violato il principio di località oppure era necessario affermare che la teoria quantistica era incompleta. In quest'ultimo caso sarebbe stato possibile ipotizzare l'esistenza di una teoria subquantica. Esisterebbero cioè delle variabili nascoste, ancora da scoprire, capaci di fornire le informazioni mancanti, permettendo così di cancellare il principio di indeterminazione e di ritornare ad una visione deterministica del mondo.
Naturalmente il gruppo di fisici mirava a dimostrare che la teoria quantistica era incompleta dal momento che il principio di località è uno dei principi fondamentali della fisica.
L’esperimento EPR è costruito su di una proprietà dei sistemi quantistici nota come entaglement. La possibilità teorica di questo fenomeno venne ipotizzata da Erwin Schrödinger nel 1926, anche se egli utilizzò per la prima volta il termine entanglement nel 1935 proprio nella recensione dell'articolo di Einstein, Podolsky e Rosen.
L'entanglement quantistico (letteralmente intreccio) o correlazione quantistica è un fenomeno che coinvolge due o più particelle generate da uno stesso processo o che si siano trovate in interazione reciproca per un certo periodo. Tali particelle rimangono in qualche modo legate indissolubilmente (entangled), nel senso che quello che accade a una di esse si ripercuote immediatamente anche sull'altra, indipendentemente dalla distanza che le separa. Il termine viene a volte reso in italiano con 'non-separabilità', in quanto uno stato entangled implica la presenza di correlazioni tra le quantità fisiche osservabili dei sistemi coinvolti.
Per esempio, è possibile realizzare un sistema entangled costituito da due particelle il cui stato quantico sia tale che - qualunque sia il valore di una certa proprietà osservabile assunto da una delle due particelle - il corrispondente valore assunto dall'altra particella sarà univocamente definito, nonostante i postulati della meccanica quantistica, secondo cui predire il risultato di queste misure sia impossibile. Di conseguenza in presenza di entanglement la misura effettuata su un sistema sembra influenzare istantaneamente lo stato di un altro sistema..
Vi sono molte versioni alternative ed equivalenti dell’esperimento EPR. In una di queste un sistema costituito di 2 particelle A e B dotate di spin ½ antiparalleli viene «preparato» in uno stato entangled da una breve interazione. Le due particelle sono poi lasciate libere di propagare verso due lontane stazioni di misura: Finchè la misura non viene effettuata ciascuna particella possiede la medesima probabilità di avere spin +½ e -½. (stati sovrapposti).
Si ipotizzi ora di misurare lo spin della particella A e di ottenere il valore + ½. Qui avviene qualcosa di assolutamente straordinario, poiché nello stesso istante la funzione d’onda della particella B subisce la riduzione: (collasso) al valore -½ con velocità dunque superiore a quella della luce, e questo senza necessità di effettuare materialmente la misura. Naturalmente se la misura dello spin di A fornisse valore -½, lo spin di B assumerebbe istantaneamente il valore +½.
E come se l’informazione ottenuta dalla misura effettuata sulla particella A producesse un’azione istantanea a distanza sulla particella B, costringendola ad assumere un particolare valore. Questo fenomeno sconcertante, sconosciuto al mondo classico, si chiama Nonlocalità Quantistica.
Da questo argomento EPR traggono la seguente conclusione: o il mondo è nonlocale (vi sono azioni in un posto che hanno ripercussioni immediate in un posto lontanissimo) oppure la meccanica quantistica non è completa. Infatti, se pensiamo che le particelle possiedano già un valore di spin ben determinato, ancorché a noi sconosciuto, scompare l’esigenza di invocare azioni a distanza ed il paradosso non è più tale.
La disuguaglianza di Bell e l’esperimento di Aspect
Solo nel 1965 John Bell, teorico del CERN, mise a punto un metodo che avrebbe potuto verificare l’esistenza o meno la presenza di effetti nonlocali in meccanica quantistica. Egli adottò i due assunti basilari di Einstein Podolsky e Rosen - l'inesistenza di segnali più veloci della luce e l'esistenza di una realtà oggettiva indipendente dalle misurazioni dello sperimentatore - e li utilizzò per costruire una relazione matematica in forma di disuguaglianza tra le misurazioni effettuate sulla particella 1 e le misurazioni effettuate sulla particella 2.
Effettuando un esperienza EPR, la disuguaglianza sarebbe stata soddisfatta nel caso l'impostazione di Einstein fosse stata corretta..
L'esperimento non poté però essere effettuato per tutti gli anni '70, poiché la tecnologia non permetteva di raggiungere i limiti di precisione richiesti.
Infatti per essere certi che due particelle separate non comunichino in modo non convenzionale (cioè istantaneamente), è necessario eseguire le misurazioni su entrambe le particelle entro un intervallo di tempo così breve che in esso nessun segnale che viaggi alla velocità della luce (o a una velocità inferiore) possa essere scambiato tra loro. Per particelle separate tra loro da una distanza di un metro, ciò significa che le misurazioni non devono impiegare più di qualche miliardesimo di secondo.
Solo nel 1982 Alain Aspect riuscì ad ottenere, in un famoso esperimento la precisione richiesta, dimostrando che Einstein aveva torto.
La nonlocalità, un monstrum scientifico secondo l’esperienza e l’intuizione umana, è ormai una proprietà generalmenteaccettata del mondo quantistico
Nella figura di seguito riportata vediamo una schematizzazione delle apparecchiature utilizzate da Aspect e collaboratori nei loro esperimenti. Al centro si trova un atomo di Calcio il cui decadimento produce una coppia di fotoni correlati che si muovono lungo percorsi opposti. Lungo uno di questi percorsi (nel caso rappresentato in figura, il Percorso A), di tanto in tanto e in maniera del tutto casuale, viene inserito un "filtro" (un Cristallo Birifrangente) il quale, una volta che un fotone interagisce con esso, può, con una probabilità del 50 %, deviarlo oppure lasciarlo proseguire indisturbato per la sua strada. Agli estremi di ogni tragitto previsto per ciascun fotone è posto un rivelatore di fotoni.
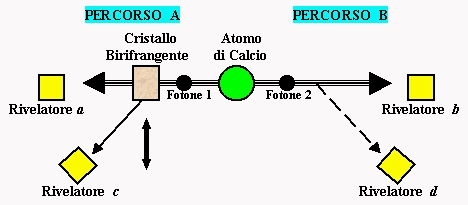
Ora, la cosa straordinaria verificata da Aspect con le sue apparecchiature è che nel momento in cui lungo il Percorso A veniva inserito il Cristallo Birifrangente e si produceva una deviazione verso il rivelatore c del fotone 1, anche il fotone 2 (ovvero il fotone del Percorso B; il fotone separato e senza "ostacoli" davanti), "spontaneamente" ed istantaneamente, deviava verso il rivelatore d. Praticamente l’atto di inserire il Cristallo Birifrangente con la conseguente deviazione del fotone 1, produceva un effetto istantaneo a distanza sul fotone 2, inducendolo a deviare.
Tutto ciò può sembrare strano, ma è quello che effettivamente accade quando si eseguono esperimenti su coppie di particelle correlate.
Conclusioni
La teoria di Newton, nella forma equivalente ma più elegante che le fu data in seguito da Hamilton, mostra che, se due corpi sono trattati come un sistema in prima approssimazione chiuso, le equazioni complete del moto possono essere dedotte dalla relazione che dà l'energia totale (potenziale + cinetica), in funzione delle masse, delle posizioni e delle quantità di moto.
In base a tale relazione, conoscendo la posizione e la quantità di moto ad un certo momento (condizioni iniziali), è sempre possibile calcolare i valori che tali grandezze assumeranno o hanno assunto in un qualsiasi momento del futuro o del passato. E tutto ciò con una precisione che dipende solamente dalla perfezione degli strumenti di misura.
In tal modo Newton introdusse nel 1687 nei suoi 'Principia Mathematica' il concetto di un sistema chiuso completamente deterministico.
Sotteso ed implicito in ciò vi era naturalmente la ferma convinzione che tale sistema esistesse ed evolvesse in modo perfettamente determinato indipendentemente dal fatto che l'uomo lo osservasse o meno. È l'assunto dell'oggettività del mondo fisico.
Fu poi Laplace a generalizzare questo concetto estendendolo all'intero universo concepito come il sistema chiuso per eccellenza, funzionante come un gigantesco meccanismo d'orologeria.
Nel suo "Theorie analytique des Probabilites" (1820), scrisse
"Un'intelligenza che conosca ad un dato istante tutte le forze agenti in natura assieme alla posizione istantanea di tutti i corpi che costituiscono l'universo è in grado di includere i moti dei maggiori corpi dell'universo e degli atomi più leggeri in una sola formula, ammesso che il suo intelletto sia sufficientemente potente da analizzare tutti i dati; niente è incerto per lui, sia passato, sia futuro sono presenti ai suoi occhi."
La meccanica quantistica ha infranto il sogno di Laplace, dimostrando che l'oggettività è un fantasma prodotto dal mondo macroscopico, ma che nel microcosmo gli oggetti esistono in modo diverso in funzione del tipo di osservazione cui sono sottoposti. Essi non hanno esistenza oggettiva, ma soggettiva, il loro mostrarsi dipende dal soggetto che li osserva.
Anche il sogno di un mondo perfettamente determinato e misurabile si è infranto contro le equazioni quantistiche. La nostra conoscenza della realtà non potrà più pretendere di essere perfetta. Dobbiamo accettare la necessità di una 'naturale’ indeterminazione, dietro la quale si nasconde una porzione di realtà attualmente per noi inconoscibile.
Nel '700 si fece strada l'idea che il caso potesse costituire l'oggetto di uno studio matematico e Laplace e altri scoprirono le leggi che governano ad esempio il gioco d'azzardo.
La cosa che forse più colpisce è che, sebbene oggi la casualità sia trattata attraverso le leggi della statistica e del calcolo delle probabilità, i matematici non riescono a dare una definizione di casualità.
Il matematico Richard von Mises ha dato una definizione operativa di un processo casuale. Secondo Von Mises, un processo è casuale se è imbattibile. Se cioè in pratica, dopo molti tentativi, qualunque strategia noi adottiamo per prevederne i risultati, i nostri sforzi risultano vani.
Se cerchiamo il caso in natura, scopriamo che il posto migliore dove trovarlo è proprio l'atomo. Non esiste casualità paragonabile a quella quantistica.
Sottoposti a controlli di casualità processi quali i decadimenti radioattivi superano ogni prova.
La casualità quantistica è imbattibile.
Il Dio che gioca a dadi non bara!
Struttura atomica e caratteristiche chimiche
Il riempimento degli orbitali
Convenzionalmente ogni orbitale viene rappresentato mediante un quadrato all'interno del quale è possibile disporre fino ad un massimo di due elettroni rappresentabili mediante frecce verticali con verso opposto, ad indicare lo spin antiparallelo.
vuoto semisaturo saturo
(elettrone spaiato)
Ciascun orbitale viene poi indicato con una sigla composta da un numero da 1 a 7 che indica il livello energetico seguito da una lettera (s, p, d, f) che indica il tipo di orbitale. Ad esempio 1s rappresenta l'unico orbitale s del primo livello energetico; 2p indica i tre orbitali p del secondo livello energetico; 6d i cinque orbitali d del sesto livello energetico.
Dato un elemento di numero atomico Z, è possibile distribuire correttamente i suoi Z elettroni nei diversi orbitali seguendo le seguenti tre regole di riempimento (Regole di Aufbau):
Principio di minima energia
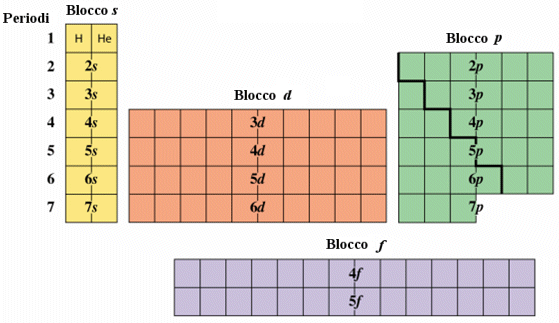
Gli elettroni si dispongono spontaneamente negli orbitali vuoti meno energetici. Una volta riempiti gli orbitali a minor energia vengono occupati gradualmente gli orbitali ad energia progressivamente maggiore. L'ordine di riempimento ottenuto in tal modo non rispetta però sempre l'ordine di riempimento che ci si attenderebbe in base alla sequenza ordinata dei livelli energetici.
Il contenuto energetico degli orbitali è riportato nello schema seguente, in cui ogni orbitale è rappresentato come un quadrato.
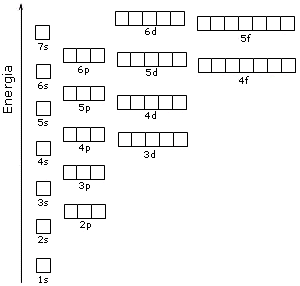
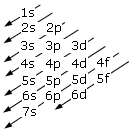
Come conseguenza di tale struttura energetica l’ordine di riempimento degli orbitali in funzione del loro contenuto energetico risulta essere quello che si ottiene seguendo le diagonali dello schema seguente.
E dunque
1s → 2s → 2p → 3s → 3p → 4s → 3d → 4p → 5s → 4d → 5p → 6s → 4f → 5d → 6p → 7s → 5f → 6d
Principio di esclusione di Pauli
Ogni orbitale può contenere al massimo 2 elettroni i quali saturano l’orbitale disponendosi con spin controversi (antiparalleli). Lo spin (momento angolare intrinseco) è una caratteristica vettoriale degli elettroni (gli elettroni si comportano come minuscole trottole che ruotano attorno al proprio asse e quindi possiedono un momento angolare). Un elettrone può possedere solo due valori di spin (anche lo spin è quantizzato). Gli elettroni negli orbitali vengono rappresentati con delle frecce verticali (che rappresentano il vettore spin).
Rappresentando dunque gli orbitali come quadrati o, come spesso si usa, come linee orizzontali, si possono presentare 3 situazioni
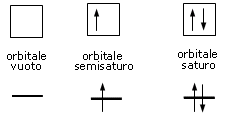
Un orbitale saturo presenta i due elettroni con spin antiparalleli (↑↓)
Principio di massima molteplicità di Hund
Gli elettroni si dispongono negli orbitali degeneri uno per orbitale con spin parallelo fino a semisaturarli tutti e, successivamente, li saturano seguendo il principio di esclusione Pauli. Così, se dobbiamo inserire 3 elettroni nei tre orbitali degeneri 2p, otterremo la seguente configurazione

Esatto Errato!!
O, dovendo inserire 7 elettroni nei cinque orbitali degeneri 4d, si otterrà
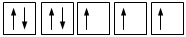
Esatto Errato!!
Applicando dunque le 3 regole di Aufbau possiamo ottenere la configurazione elettronica dell’Ossigeno (Z = 8). I suoi 8 elettroni si distribuiranno secondo il seguente schema

La configurazione elettronica dell’Ossigeno può essere riassunta in modo sintetico scrivendo gli elettroni ad esponente degli orbitali che li contengono
1s2 2s2 2p4
in cui i numeri ad esponente indicano quanti elettroni sono sistemati in quei particolari orbitali.
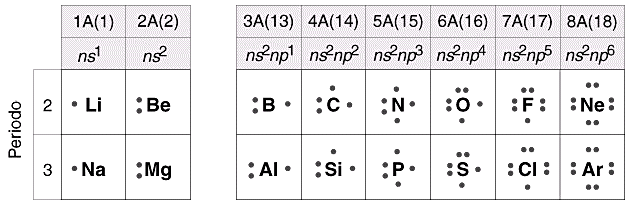
Configurazioni elettroniche e Strutture di Lewis degli elementi
Se prendiamo in considerazione i diversi elementi a partire dall'idrogeno in ordine di numero atomico crescente possiamo osservare come al crescere di un'unità nel valore del numero atomico venga aggiunto un elettrone alla configurazione elettronica.
Ogniqualvolta una serie di elementi ha sistemato abbastanza elettroni da riempire un livello energetico, gli elementi successivi, che iniziano a riempire il successivo livello energetico, vengono disposti in una riga sottostante, detta periodo, in modo tale che risultino incolonnati con gli elementi che presentano la stessa configurazione elettronica superficiale.
In questo modo si vengono a formare 7 periodi, corrispondenti ai 7 livelli energetici riempibili e quindi al valore del numero quantico principale.
Gli elementi che si incolonnano verticalmente formano i cosiddetti gruppi, composti da elementi che presentano un egual numero di elettroni disposti sullo stesso tipo di orbitali, ma naturalmente su di un diverso livello energetico.
Poiché il comportamento chimico di un elemento dipende essenzialmente proprio dalla sua configurazione elettronica superficiale, elementi appartenenti ad uno stesso gruppo presentano forti analogie e somiglianze chimiche (stesso tipo di reazioni).
Le caratteristiche chimiche variano dunque progressivamente e con continuità mentre ci spostiamo lungo un periodo, mentre rimangono sostanzialmente simili all'interno di un gruppo.
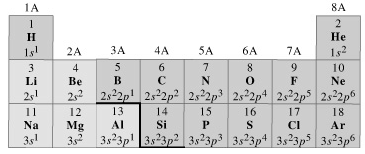
Possiamo inoltre suddividere la tabella periodica in quattro grandi regioni che rappresentano raggruppamenti di elementi che stanno inserendo elettroni in orbitali dello stesso tipo.
La prima regione (blocco s) è formata dai gruppi I A e II A dove si riempie l'orbitale s.
La regione all'estrema destra (blocco p), composta dai rimanenti sei gruppi A (l’ottavo gruppo A è spesso indicato come gruppo 0 (zero)) è costituita dagli elementi che distribuiscono i sei elettroni nei tre orbitali p.
La regione centrale (blocco d), costituita da 10 file verticali riunite a formare 8 gruppi B, è formata dai cosiddetti metalli di transizione, elementi che stanno disponendo 10 elettroni nei 5 orbitali d.
Infine la regione in basso (blocco f), formata da due serie orizzontali chiamate rispettivamente serie dei lantanidi e degli attinidi, è costituita da elementi che stanno distribuendo 14 elettroni nei 7 orbitali f (4f e 5f).
Gli atomi utilizzano prevalentemente gli elettroni del loro livello energetico più esterno (elettroni superficiali o elettroni di valenza) per interagire e legarsi tra loro. Il comportamento chimico di un atomo dipende dunque dal numero e dalla disposizione degli elettroni dell’ultimo livello energetico. Per capire la reattività di un atomo è dunque sufficiente conoscere la sua configurazione elettronica superficiale o configurazione dello strato di valenza.
Scrivendo, ad esempio, le configurazioni elettroniche superficiali degli elementi dei primi tre periodi chimici della tabella periodica, si osserva come gli elementi che si incolonnano in uno stesso gruppo chimico presentano la medesima configurazione elettronica superficiale.
La configurazione elettronica superficiale semplicemente si ripete periodicamente in livelli energetici via via più esterni.
Atomi di elementi diversi che presentino la medesima configurazione elettronica superficiale (il medesimo numero di elettroni sul loro ultimo livello) manifestano caratteristiche chimiche simili.
Così, ad esempio, lo Zolfo, che si trova sotto l’Ossigeno, presenta la medesima configurazione superficiale (ns2 np4) di quest’ultimo, sul terzo livello energetico (n = 3) invece che sul secondo (n = 2). Per questo motivo Zolfo ed Ossigeno hanno caratteristiche chimiche simili.
Dunque le caratteristiche simili degli elementi che appartengono ad uno stesso gruppo chimico dipendono essenzialmente dal numero di elettroni presenti sul livello energetico più superficiale, indipendentemente dal fatto che questo sia il primo, il secondo o l'ultimo.
Il numero d'ordine di ciascun gruppo indica quanti elettroni sono presenti nel livello energetico superficiale, dandoci quindi una prima indicazione di massima sul numero di elettroni disponibili per i legami chimici.
Così tutti gli elementi del primo gruppo A presentano configurazione elettronica superficiale ns1, dove n indica evidentemente il numero quantico principale.
Possiamo dunque costruire il seguente schema che ci permette di correlare ciascun gruppo A con la configurazione elettronica superficiale degli elementi appartenenti al gruppo stesso.
gruppo |
configurazione |
numero |
I A |
ns1 |
1 |
II A |
ns2 |
2 |
III A |
ns2 np1 |
3 |
IV A |
ns2 np2 |
4 |
V A |
ns2 np3 |
5 |
VI A |
ns2 np4 |
6 |
VII A |
ns2 np5 |
7 |
VIII A |
ns2 np6 |
8 |
Gli elementi che possiedono 8 elettroni superficiali (configurazione otteziale) risultano particolarmente stabili, inerti, nel senso che manifestano pochissima tendenza a reagire con altri elementi chimici.
Gli altri elementi che possiedono configurazioni elettroniche simili a quella dei gas nobili tendono a perdere o ad acquistare elettroni per raggiungere tale configurazione particolarmente stabile. Molte reazioni chimiche possono essere spiegate proprio in virtù della tendenza di molti elementi ad acquisire la configurazione ad 8 elettroni superficiali dei gas nobili (regola dell'ottetto).
Spesso la configurazione elettronica di un elemento viene scritta in forma sintetica facendo riferimento al gas nobile che lo precede nella tabella periodica, e che presenta tutti i suoi livelli energetici completi, ed aggiungendo solo la configurazione superficiale dell’elemento.
Ad esempio le configurazioni di Ossigeno e Zolfo possono essere scritte così
Ossigeno 1s2 2s2 2p4 = [He]2s22p4
Zolfo 1s2 2s2 2p6 3s2 3p4 = [Ne]3s23p4
In altre parole, l’Ossigeno presenta la stessa configurazione elettronica dell’Elio più la sua configurazione superficiale, mentre lo Zolfo presenta la medesima configurazione elettronica del Neon più la sua configurazione superficiale.
I gruppi B vengono ordinati in analogia ai gruppi A. Il primo gruppo B che si forma viene detto III B e non I B in quanto la sua configurazione superficiale presenta 3 elettroni, 2 nell'orbitale s e 1 nell'orbitale d, in modo analogo a quanto avviene per gli elementi del gruppo III A.
Tranne alcune eccezioni anche per i gruppi B vale la regola che il numero d'ordine indica il numero di elettroni presenti nello strato più superficiale.
I lantanidi e gli attinidi hanno caratteristiche chimiche simili rispettivamente al lantanio e all'attinio e si suole perciò considerarli appartenenti al gruppo III B.
Nella maggior parte dei casi gli elettroni coinvolti nelle reazioni chimiche sono i 2 + 6 = 8 elettroni contenuti nell’orbitale s e nei tre orbitali p del livello energetico più esterno. Per questo motivo tali elettroni vengono convenzionalmente rappresentati utilizzando un metodo introdotto da Lewis.
Secondo tale metodo i 2 + 6 elettroni degli orbitali s e p del livello più esterno (elettroni di valenza) vengono rappresentati come punti o coppie di punti disposte ai quattro lati del simbolo chimico dell'elemento. Idealmente, ogni lato del simbolo chimico è associato ad un orbitale.
Per maggior chiarezza diamo la configurazione di Lewis o struttura di Lewis degli elementi appartenenti al 2° periodo.
Elemento |
Orbitale s |
Orbitali p |
Configurazione |
Configurazione |
Litio |
|
|
2s1 |
|
Berillio |
|
|
2s2 |
|
Boro |
|
|
2s22p1 |
|
Carbonio |
|
|
2s22p2 |
|
Azoto |
|
|
2s22p3 |
|
Ossigeno |
|
|
2s22p4 |
|
Fluoro |
|
|
2s22p5 |
|
Neon |
|
|
2s22p6 |
|
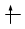
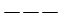
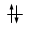
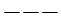
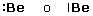
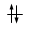
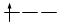
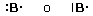
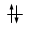
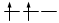
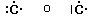
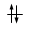
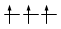
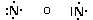
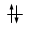
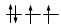
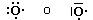
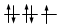
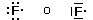
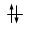
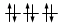
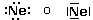
Come si vede, le coppie di elettroni vengono più spesso rappresentate con una barretta.
E’ evidente che tutti gli elementi che appartengono ad un medesimo gruppo chimico, possedendo la medesima configurazione elettronica superficiale, presentano la stessa struttura di Lewis
Metalli e non metalli
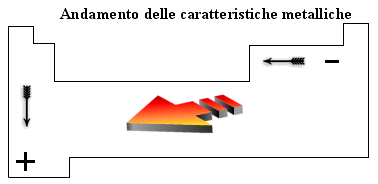
E' possibile tracciare all'interno della tabella periodica una ideale linea obliqua che, passando per il Silicio (Si), l'Arsenico (As) ed il Tellurio (Te), va dal Boro (B) all'Astato (At) e divide tutti gli elementi in due grandi gruppi: a sinistra i metalli (più numerosi), a destra i non metalli. Le caratteristiche chimiche e fisiche dei metalli sono più accentuate all'inizio della tabella periodica e vanno lentamente sfumando mentre ci avviciniamo alla zona dei non metalli.
Gli elementi chimici che si trovano adiacenti alla linea di separazione presentano quindi caratteristiche intermedie tra quelle dei metalli e quelle dei non metalli e vengono per questo motivo chiamati semi-metalli. I metalli presentano una tendenza a perdere elettroni (si ossidano più o meno facilmente) trasformandosi in ioni positivi o cationi. Dal punto di vista fisico sono lucenti, tenaci (si rompono con difficoltà), duttili ( possono essere tirati in fili sottili), malleabili (possono essere tirati in lamine sottili), buoni conduttori di calore e di elettricità.
I non metalli presentano una tendenza ad acquistare elettroni (si riducono più o meno facilmente) trasformandosi in ioni negativi o anioni. Dal punto di vista fisico non sono lucenti, sono fragili, non presentano né duttilità, né malleabilità, sono cattivi conduttori o addirittura isolanti termici ed elettrici.
Le caratteristiche metalliche aumentano scendendo lungo un gruppo e spostandosi verso sinistra lungo un periodo. In tal modo gli elementi che presentano le caratteristiche metalliche più spiccate sono quelli in basso a sinistra della tabella periodica.
Evidentemente per ragioni opposte gli elementi che presentano le caratteristiche non metalliche più accentuate si trovano in alto a destra nella tabella periodica
Possiamo trovare una semplice spiegazione di tale andamento analizzando come varia il raggio atomico. Osservando la tabella periodica è facile verificare che il raggio atomico, e quindi la distanza degli elettroni più superficiali dal loro nucleo, diminuisce da sinistra verso destra lungo un periodo mentre aumenta dall'alto in basso lungo un gruppo.
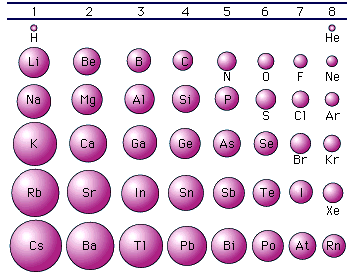
Infatti mentre ci spostiamo verso destra lungo un periodo gli elettroni vengono sistemati tutti in uno stesso livello energetico. La distanza di tale livello dal nucleo dovrebbe rimanere approssimativamente la stessa. In realtà poiché contemporaneamente aumenta anche il numero atomico Z, il nucleo esercita una attrazione via via maggiore sui livelli energetici occupati dagli elettroni, costringendoli a contrarsi verso il centro.
Quando invece ci muoviamo verso il basso lungo un gruppo ciascun elemento presenta i suoi elettroni superficiali su livelli energetici nuovi e via via più esterni, facendo in tal modo aumentare di scatto il raggio atomico
Ora è evidente che più distanti gli elettroni superficiali si trovano dal nucleo positivo e minore è la forza attrattiva che il nucleo stesso è in grado di esercitare su di essi. Ciò spiega in definitiva la maggior facilità con cui gli atomi metallici, che possiedono raggi atomici mediamente superiori rispetto ai non metalli, perdono i loro elettroni superficiali.
La tendenza a perdere elettroni da parte dei metalli è inoltre esaltata dal fatto che i metalli possiedono in genere pochi elettroni in più rispetto al gas nobile che li precede ed è per loro energeticamente più conveniente perderli piuttosto che acquistare un gran numero di elettroni per raggiungere la configurazione stabile del gas nobile che li segue nella tabella periodica.
Per ragioni opposte per i non metalli, che presentano in genere pochi elettroni in meno rispetto al gas nobile che li segue, è energeticamente più favorevole acquistarli piuttosto che perderne un gran numero.
La tendenza a perdere o ad acquistare elettroni da parte degli elementi chimici è misurata da due parametri fondamentali, l'energia di I ionizzazione e l'affinità elettronica, i cui valori si trovano tabulati nella tabella periodica.
Energia di prima ionizzazione
Viene definita come l'energia, espressa in Kcal/mol (o KJ/mol), che è necessario fornire ad una mole di atomi allo stato gassoso per trasformarla in una mole di cationi monovalenti.
X(g) + EI ion (kJ/mol) → X+(g) + e-
L'andamento del valore di tale parametro nella tabella periodica ci conferma quanto abbiamo detto sulla maggior facilità con la quale i metalli perdono i loro elettroni.
L'energia di ionizzazione diminuisce infatti scendendo verso il basso lungo un gruppo, mentre cresce se ci spostiamo verso destra lungo un periodo.
Energia di Prima Ionizzazione (kJ mol-1) |
||||||||||||||||||
1 |
H |
|
He |
|||||||||||||||
2 |
Li |
Be |
|
B |
C |
N |
O |
F |
Ne |
|||||||||
3 |
Na |
Mg |
|
Al |
Si |
P |
S |
Cl |
Ar |
|||||||||
4 |
K |
Ca |
Sc |
Ti |
V |
Cr |
Mn |
Fe |
Co |
Ni |
Cu |
Zn |
Ga |
Ge |
As |
Se |
Br |
Kr |
5 |
Rb |
Sr |
Y |
Zr |
Nb |
Mo |
Tc |
Ru |
Rh |
Pd |
Ag |
Cd |
In |
Sn |
Sb |
Te |
I |
Xe |
6 |
Cs |
Ba |
La |
Hf |
Ta |
W |
Re |
Os |
Ir |
Pt |
Au |
Hg |
Tl |
Pb |
Bi |
Po |
At |
Rn |
7 |
Fr |
Ra |
Ac |
Rf |
Db |
Sg |
Bh |
Hs |
Mt |
Ds |
Rg |
Uub |
Uut |
Uuq |
Uup |
Uuh |
Uus |
Uuo |
Ce 527 |
Pr |
Nd |
Pm |
Sm |
Eu |
Gd |
Tb |
Dy |
Ho |
Er |
Tm |
Yb |
Lu |
Th |
Pa |
U |
Np |
Pu |
Am |
Cm |
Bk |
Cf |
Es |
Fm |
Md |
No |
Lr |
Affinità elettronica
Viene definita come l'energia liberata da una mole di atomi neutri allo stato gassoso quando si trasforma in una mole di anioni monovalenti.
X(g) + e- → X-(g) + AE (kJ/mol)
Tale definizione è contraria alla convenzione secondo la quale l’energia liberata durante una reazione ha segno negativo, generando spesso non poca confusione. Per questo motivo a volte si preferisce definire l’affinità elettronica come l’energia di ionizzazione degli ioni negativi, cioè come l’energia che deve essere fornita (quindi con segno positivo) ad uno ione negativo per strappargli il suo elettrone
X-(g) + EAE (kJ/mol) → X(g) + e-
L'andamento dei valori dell'affinità elettronica è analogo a quello del potenziale di Ia ionizzazione. Cresce lungo un periodo e decresce lungo un gruppo.
Affinità Elettronica (kJ mol-1) |
||||||||||||||||||
1 |
H |
|
He |
|||||||||||||||
2 |
Li |
Be |
|
B |
C |
N |
O |
F |
Ne |
|||||||||
3 |
Na |
Mg |
|
Al |
Si |
P |
S |
Cl |
Ar |
|||||||||
4 |
K |
Ca |
Sc |
Ti |
V |
Cr |
Mn |
Fe |
Co |
Ni |
Cu |
Zn |
Ga |
Ge |
As |
Se |
Br |
Kr |
5 |
Rb |
Sr |
Y |
Zr |
Nb |
Mo |
Tc |
Ru |
Rh |
Pd |
Ag |
Cd |
In |
Sn |
Sb |
Te |
I |
Xe |
6 |
Cs |
Ba |
La |
Hf |
Ta |
W |
Re |
Os |
Ir |
Pt |
Au |
Hg |
Tl |
Pb |
Bi |
Po |
At |
Rn |
7 |
Fr |
Ra |
Ac |
Rf |
Db |
Sg |
Bh |
Hs |
Mt |
Ds |
Rg |
Uub |
Uut |
Uuq |
Uup |
Uuh |
Uus |
Uuo |
Altre informazioni utili nella tabella periodica
- numero atomico Z = numero di protoni presenti nel nucleo
- configurazione elettronica
- peso atomico relativo espresso in uma (o dalton) = rapporto tra il peso di un elemento (miscela dei suoi isotopi) ed 1/12 della massa del carbonio 12. Ricordiamo inoltre che il peso atomico relativo è numericamente pari al peso molare (PM) dell'elemento stesso.
Altre informazioni utili ottenibili dalla consultazione della tabella sono quelle relative al numero di ossidazione e all'elettronegatività degli elementi, di cui parleremo in seguito.
I legami chimici
Gli atomi tendono a raggiungere delle configurazioni energeticamente più favorevoli e stabili legandosi in raggruppamenti (molecole, reticolati ionici, reticolati metallici). In altre parole gli atomi si legano perché il composto che ne deriva è più stabile degli atomi separati.
La teoria del legame chimico si fonda, nella sua formulazione più semplice, sulla legge di Coulomb.
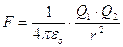
dove q sono le cariche elettriche, r la distanza che le separa ed F la forza (attrattiva per cariche opposte, repulsiva per cariche concordi) che si esercita su di esse.
Quando due atomi vengono avvicinati le nuvole elettroniche ed i nuclei interagiscono tra loro. Il legame è il risultato di un bilanciamento tra forze coulombiane attrattive (elettroni-nuclei) e repulsive (elettroni-elettroni e nuclei-nuclei).

Tuttavia tali forze risultano essere di intensità diversa per i diversi atomi e ciò porta alla formazione di legami con modalità differenti. Esistono tre modelli di legame (covalente, ionico, metallico) le cui caratteristiche dipendono essenzialmente dalla tendenza relativa che manifestano gli atomi coinvolti nel legame ad acquistare (affinità elettronica) o perdere elettroni (energia di ionizzazione).
Il legame covalente si presenta tipicamente tra atomi con elevata affinità elettronica (atomi di elementi non metallici)
Il legame ionico si presenta tipicamente tra atomi con elevata affinità elettronica (non metalli) ed atomi a bassa energia di ionizzazione (metalli)
Il legame metallico si presenta tipicamente tra atomi di elementi con bassa energia di ionizzazione (metalli)
Poiché, come abbiamo già detto, gli elettroni coinvolti nei legami chimici sono quelli che occupano il livello energetico più superficiale (elettroni di valenza), introduciamo un metodo semplice per rappresentarli, noto come configurazione di Lewis degli elementi.
Il legame covalente: Teoria di Lewis
Il legame covalente si forma tra atomi che presentano alta affinità elettronica e quindi tipicamente tra atomi non metallici.
Se il legame unisce atomi di un medesimo elemento, il legame si definisce covalente puro o covalente omeopolare.
Se il legame unisce atomi di elementi diversi, il legame si definisce covalente polare o covalente eteropolare.
La natura del legame covalente venne suggerita per la prima volta da G. Lewis, dell'università della California nel 1916.
Lewis attribuì l'inerzia chimica dei gas nobili al fatto di possedere 8 elettroni superficiali e avanzò quindi l'ipotesi che gli elementi che non presentavano la stessa configurazione elettronica esterna, tendessero a raggiungerla mediante la condivisione dei loro elettroni superficiali spaiati, al fine di raggiungere in tal modo una configurazione più stabile (regola dell’ottetto).
Prendiamo ad esempio due atomi di cloro, rappresentandoli mediante le loro strutture di Lewis. Essi hanno entrambi una configurazione 3s2 3p5, con un elettrone spaiato sull'ultimo orbitale p ed una forte tendenza ad acquistare un ulteriore elettrone (elevata affinità elettronica) per raggiungere la configurazione stabile del gas nobile successivo ( l'argon).
Possiamo pensare che entrambi i nuclei attirino fortemente l'elettrone spaiato dell'altro atomo senza peraltro riuscire a strapparlo.
Il risultato di questa intensa attrazione incrociata è che i due elettroni spaiati vengono alla fine condivisi da entrambi gli atomi ed il doppietto elettronico funge da legame, finendo per appartenere ad entrambi gli atomi.
I due atomi di Cloro “condividono” una coppia di elettroni e tale “condivisione” costituisce il legame covalente. In questo modo ora i due elettroni non appartengono più all'uno o all'altro atomo, ma ruotano entrambi intorno all'intera struttura molecolare biatomica.
Si dice che i due elettroni sono stati messi in comune o in compartecipazione.
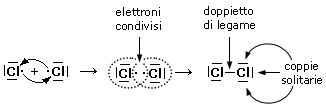
Ciascun nucleo "vede" ora intorno a sè i 6 elettroni non condivisi più i 2 elettroni condivisi per un totale di 8 elettroni. La condivisione di una coppia di elettroni permette a ciascun atomo di Cloro di raggiungere la configurazione stabile dell’ottetto.
Il legame che si forma per condivisione di una coppia di elettroni è detto legame covalente semplice o singolo e può essere rappresentato mediante una barretta che unisce i due simboli chimici. Gli atomi che formano la molecola di Cl2 sono quindi tenuti insieme da un legame covalente semplice
Cl - Cl
Le coppie di elettroni superficiali che non vengono condivise sono dette coppie (o doppietti) di non-legame o coppie solitarie (Lone Pairs).
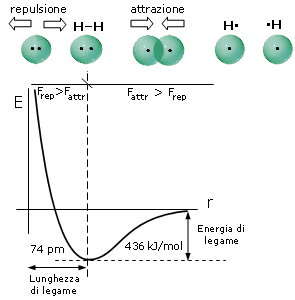
Come abbiamo già detto, durante il processo di formazione del legame si esercitano tra i due atomi sia forze attrattive (elettroni-nuclei) che forze repulsive (tra i nuclei, ma soprattutto tra i gusci elettronici). Le forze attrattive prevalgono a distanze maggiori, consentendo in questo modo ai due atomi di avvicinarsi e legarsi, mentre le forze repulsive diventano importanti e significative a corto raggio. L’energia potenziale del sistema (convenzionalmente posta uguale a zero quando i due atomi si trovano idealmente a distanza infinita) diminuisce man mano che le forze attrattive costringono i due atomi ad avvicinarsi.
Via via che i due atomi si avvicinano sia le forze attrattive che quelle repulsive diventano progressivamente più intense. Tuttavia, poiché le forze repulsive aumentano più rapidamente di quelle attrattive, si arriverà ad una distanza critica 8distanza di legame) in corrispondenza della quale le due forze risulteranno perfettamente uguali, l’energia potenziale raggiungerà il suo valore minimo ed il sistema sarà in equilibrio. Ogni ulteriore avvicinamento degli atomi causerà un aumento delle forze repulsive maggiore di quelle attrattive con conseguente tendenza del sistema a ritornare alla distanza di equilibrio.
Si tenga presente che la pendenza della curva dell’energia potenziale, rappresenta la forza netta (attrattiva + repulsiva) che agisce sugli atomi.
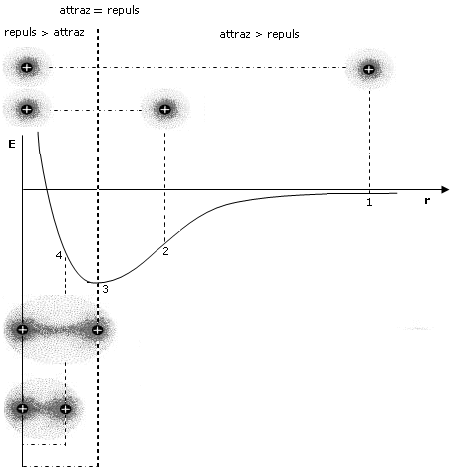
A grandi distanze (punto 1 del grafico) la curva dell’energia potenziale presenta piccole pendenze positive che corrispondono a deboli forze nette attrattive. Via via che gli atomi si avvicinano la pendenza della curva aumenta fino a raggiungere un valore massimo (punto 2 del grafico) in corrispondenza del quale le forze repulsive iniziano a crescere più rapidamente di quelle attrattive e la forza netta di attrazione inizia a diminuire. Alla distanza di legame (punto 3 del grafico) l’energia potenziale raggiunge il suo valore minimo e la forza netta è pari a zero (pendenza nulla e forze attrattive pari a quelle repulsive). A distanze inferiori (punto 4 del grafico) la curva dell’energia potenziale presenta pendenze negative crescenti che corrispondono ad intense forze repulsive nette.
Si può dunque dimostrare che, quando due atomi si avvicinano in risposta all'attrazione che ciascun nucleo esercita sull'elettrone spaiato dell'altro atomo, esiste una distanza critica in corrispondenza della quale la forza di attrazione viene esattamente bilanciata dalla repulsione che si produce tra i gusci elettronici negativi. Per distanze inferiori prevale la repulsione, per distanze maggiori prevale l'attrazione. Un modello semplice ed intuitivo, che descrive il fenomeno, rappresenta il legame come una molla che unisce i due atomi. Se si cerca di separarli la molla li richiama, se si cerca di avvicinarli troppo la molla li respinge, alla distanza di legame la molla non è in tensione.
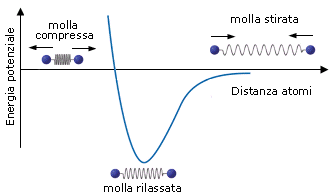
In corrispondenza della distanza di equilibrio viene quindi resa minima l'energia potenziale del sistema. Tale distanza corrisponde alla lunghezza di legame, parametro solitamente misurato in Ǻngström (1Ǻ = 10-10 m) o in picometri (1 pm = 10-12 m).
L'energia di legame, misurata in Kcal/mol (o in kJ/mol), è l’energia che si libera quando due atomi allo stato gassoso passano da distanza infinita alla distanza di legame ed ovviamente coincide con l'energia che è necessario fornire al sistema (allo stato gassoso) per rompere il legame, portando i due atomi a distanza infinita.
X-X(g) + Eleg (kcal/mol) → X•(g) + X•(g)
L’energia di legame è una misura della “forza” di un legame chimico (maggiore è l’energia di legame, più “forte” è un legame) e per questo motivo viene a volte impropriamente detta forza di legame.
Si consideri ad esempio la formazione del legame covalente in una molecola di H2.
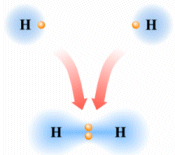
Due atomi di idrogeno condividono il loro unico elettrone per raggiungere la configurazione stabile dell’Elio.
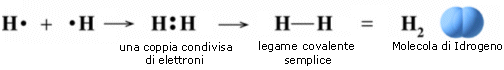
Nello schema successivo è rappresentato l’andamento dell’energia potenziale durante la formazione del legame.
Energia E (in kJ/mol) e Lunghezza L (in pm) di legame |
||||||||||||||||||
|
E |
L |
|
|
E |
L |
|
|
E |
L |
|
|
E |
L |
|
|
E |
L |
H-H |
432 |
74 |
N-Cl |
313 |
175 |
C-Pb |
130 |
230 |
Ge-Ge |
188 |
241 |
S-S (S8) |
226 |
205 |
||||
H-B |
389 |
119 |
P-P |
201 |
221 |
C-N |
305 |
147 |
Ge-N |
257 |
|
S=S |
425 |
149 |
||||
H-C |
411 |
109 |
P-O |
335 |
163 |
C=N |
615 |
129 |
Ge-F |
470 |
168 |
S-F |
284 |
156 |
||||
H-Si |
318 |
148 |
P=O |
544 |
150 |
C≡N |
887 |
116 |
Ge-Cl |
349 |
210 |
S-Cl |
255 |
207 |
||||
H-Ge |
288 |
153 |
P=S |
335 |
186 |
C-P |
264 |
184 |
Ge-Br |
276 |
230 |
Se-Se |
172 |
|
||||
H-Sn |
251 |
170 |
P-F |
490 |
154 |
C-O |
358 |
143 |
Ge-I |
212 |
|
Se=Se |
272 |
215 |
||||
H-N |
386 |
101 |
P-Cl |
326 |
203 |
C=O |
799 |
120 |
Sn-F |
414 |
|
F-F |
155 |
142 |
||||
H-P |
322 |
144 |
P-Br |
264 |
|
C≡O |
1072 |
113 |
Sn-Cl |
323 |
233 |
Cl-Cl |
240 |
199 |
||||
H-As |
247 |
152 |
P-I |
184 |
|
C-B |
356 |
|
Sn-Br |
273 |
250 |
Br-Br |
190 |
228 |
||||
H-O |
459 |
96 |
As-As |
146 |
243 |
C-S |
272 |
182 |
Sn-I |
205 |
270 |
I-I |
148 |
267 |
||||
H-S |
363 |
134 |
As-O |
301 |
178 |
C=S |
573 |
160 |
Pb-F |
331 |
|
At-At |
116 |
|
||||
H-Se |
276 |
146 |
As-F |
484 |
171 |
C-F |
485 |
135 |
Pb-Cl |
243 |
242 |
I-O |
201 |
|
||||
H-Te |
238 |
170 |
As-Cl |
322 |
216 |
C-Cl |
327 |
177 |
Pb-Br |
201 |
|
I-F |
273 |
191 |
||||
H-F |
565 |
92 |
As-Br |
458 |
233 |
C-Br |
285 |
194 |
Pb-I |
142 |
279 |
I-Cl |
208 |
232 |
||||
H-Cl |
428 |
127 |
As-I |
200 |
254 |
C-I |
213 |
214 |
B-B |
293 |
|
I-Br |
175 |
|
||||
H-Br |
362 |
141 |
Sb-Sb |
121 |
|
Si-Si |
222 |
233 |
B-O |
536 |
|
Xe-O |
84 |
175 |
||||
H-I |
295 |
161 |
Sb-F |
440 |
|
Si-N |
355 |
|
B-F |
613 |
|
Xe-F |
130 |
195 |
||||
N-N |
167 |
145 |
C-C |
346 |
154 |
Si-O |
452 |
163 |
B-Cl |
456 |
175 |
|
|
|
||||
N=N |
418 |
125 |
C=C |
602 |
134 |
Si-S |
293 |
200 |
B-Br |
377 |
|
|
|
|
||||
N≡N |
942 |
110 |
C≡C |
835 |
120 |
Si-F |
565 |
160 |
O-O |
142 |
148 |
|
|
|
||||
N-O |
201 |
140 |
C-Si |
318 |
185 |
Si-Cl |
381 |
202 |
O=O |
494 |
121 |
|
|
|
||||
N=O |
607 |
121 |
C-Ge |
238 |
195 |
Si-Br |
310 |
215 |
O-F |
190 |
142 |
|
|
|
||||
N-F |
283 |
136 |
C-Sn |
192 |
216 |
Si-I |
234 |
243 |
S=O |
522 |
143 |
|
|
|
||||
Nella formazione di un legame covalente possono essere condivise anche più di una coppia di elettroni. E' il caso ad esempio delle molecole dell'ossigeno e dell'azoto.
L'ossigeno presenta 6 elettroni nell'ultimo livello con una configurazione elettronica superficiale 2s2 2p4, con due elettroni spaiati su due orbitali p. Per completare l'ottetto ciascun atomo di ossigeno condivide dunque 2 elettroni. Ciascun atomo di ossigeno ora "vede" intorno a sè 8 elettroni nel suo livello energetico più superficiale.
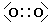
La molecola di O2 è quindi tenuta insieme da un legame covalente doppio. che può essere rappresentato con due trattini posti tra i simboli chimici dei due atomi (O = O)
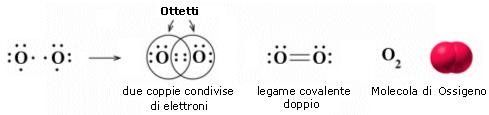
Un legame doppio è più corto e più forte di un legame semplice.
Nel caso dell'azoto la configurazione elettronica superficiale è del tipo 2s2 2p3, con tre elettroni spaiati su due orbitali p. Per completare l'ottetto ciascun atomo di Azoto deve dunque condividere 3 elettroni
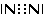
Il legame che si forma e che tiene uniti gli atomi di Azoto in N2 è un legame covalente triplo che può essere rappresentato con tre trattini posti tra i simboli chimici dei due atomi (N ºN).

Un legame triplo è più corto e più forte di un legame doppio.
Riassumendo
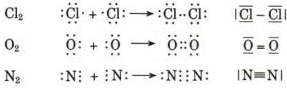
Il numero di doppietti elettronici condivisi che tiene uniti due atomi è detto ordine di legame. Un legame singolo presenta ordine = 1, un legame doppio ordine = 2, un legame triplo ordine = 3.
La lunghezza del legame decresce all’aumentare dell’ordine di legame (un legame doppio è più corto di uno semplice ed uno triplo è più corto di uno doppio).
L’energia di legame aumenta all’aumentare dell’ordine di legame (un legame doppio è più forte di un legame semplice ed un legame triplo è più forte di un legame doppio).
Mentre i legami semplici permettono la libera rotazione degli atomi intorno all'asse di legame, i legami doppi e tripli non permettono rotazioni.
Raggio covalente
La lunghezza dei legami covalenti è correlabile al raggio covalente degli elementi.
Si definisce raggio covalente di un elemento la metà della lunghezza del legame covalente singolo che tiene uniti due atomi del medesimo elemento in una molecola neutra. In realtà tale definizione non si può applicare a tutti gli elementi.
L’ossigeno, ad esempio, forma una molecola O2 in cui i due atomi sono uniti da un legame di ordine = 2 (covalente doppio e non singolo). In questo caso si stima il raggio covalente analizzando molecole che contengano il gruppo -O-O-.
In altri casi si misura la lunghezza del legame A-X che un elemento X presenta con l’elemento A di cui è noto il raggio covalente e si stima il suo raggio covalente per differenza con il raggio covalente noto di A. Spesso si usa il Carbonio che si lega facilmente con molti elementi chimici e di cui è noto il raggio covalente.
Nel caso di elementi metallici, i cui atomi sono tenuti insieme da un legame metallico, si parla più propriamente di raggio metallico. Più in generale si parla di raggio atomico, covalente o metallico, in relazione al tipo di legame che tiene uniti gli atomi.
Raggio atomico (covalente e metallico) (in pm) |
|||||||||||||||||
H |
|
He |
|||||||||||||||
Li |
Be |
|
B |
C |
N |
O |
F |
Ne |
|||||||||
Na |
Mg |
|
Al |
Si |
P |
S |
Cl |
Ar |
|||||||||
K |
Ca |
Sc |
Ti |
V |
Cr |
Mn |
Fe |
Co |
Ni |
Cu |
Zn |
Ga |
Ge |
As |
Se |
Br |
Kr |
Rb |
Sr |
Y |
Zr |
Nb |
Mo |
Tc |
Ru |
Rh |
Pd |
Ag |
Cd |
In |
Sn |
Sb |
Te |
I |
Xe |
Cs |
Ba |
La |
Hf |
Ta |
W |
Re |
Os |
Ir |
Pt |
Au |
Hg |
Tl |
Pb |
Bi |
Po |
At |
Rn |
Fr |
Ra |
Ac |
Rf |
Db |
Sg |
Bh |
Hs |
Mt |
Ds |
Rg |
Uub |
Uut |
Uuq |
Uup |
Uuh |
Uus |
Uuo |
Legame dativo e promozione elettronica
Da quanto abbiamo fin qui visto ci si potrebbe attendere che il numero di legami covalenti che un atomo può formare non possa mai essere superiore al numero di elettroni spaiati che deve condividere al fine di completare l'ottetto. In realtà ciò è vero nella maggior parte dei casi, ma non in tutti.
In alcuni casi gli atomi riescono infatti a formare più legami di quanti apparentemente sembra loro consentito sulla base della loro configurazione elettronica superficiale. E' necessario tener presente che ogni legame in più che si forma produce un ulteriore aumento di stabilità della molecola e dunque gli atomi tendono a massimizzare il numero di legami formati.
La formazione di legami di tipo “dativo” ed i processi di “promozione elettronica” sono stati introdotti per giustificare la formazione di ulteriori legami rispetto al numero di elettroni spaiati di un atomo.
Legame dativo
Un legame covalente si può formare a partire da un doppietto elettronico messo a disposizione da un atomo donatore (o datore) e da un orbitale vuoto messo a disposizione da un atomo accettore.
Tale legame è detto legame covalente dativo ed una volta formatosi è indistinguibile da un normale legame covalente.
Il legame dativo può essere rappresentato come una freccia che va dal doppietto solitario dell’atomo datore D all’atomo accettore A.
D:→A
Si consideri ad esempio lo Zolfo e l'Ossigeno, entrambi appartenenti al VI gruppo A, aventi configurazione superficiale s2p4, con due doppietti, due elettroni spaiati e due elettroni mancanti per raggiungere la configurazione dell'ottetto.
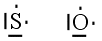
Sulla base delle configurazioni superficiali dei due elementi ci potremmo attendere la formazione di un composto del tipo S=O, con formazione di un legame covalente doppio a seguito della condivisione di entrambi gli elettroni spaiati di ciascun elemento. In questo modo ciascun atomo completa infatti l’ottetto.

In realtà quando lo Zolfo reagisce con l’Ossigeno genera SO2 (anidride solforosa) ed SO3 (anidride solforica), riuscendo in tal modo a legare fino ad altri due atomi di ossigeno in più rispetto a quanto previsto.
Per giustificare la formazione di questi legami si ammette dunque che lo zolfo possa utilizzare per legarsi non solo gli elettroni spaiati, ma anche i doppietti solitari. Tuttavia l’Ossigeno non ha orbitali superficiali vuoti da poter utilizzare.
Si ammette quindi che l'Ossigeno possa subire una transizione dalla configurazione più stabile, prevista dalla regola di Hund ad una configurazione, meno stabile nella quale un elettrone viene spostato da un orbitale p semisaturo, generando un orbitale vuoto, all’altro orbitale p semisaturo.
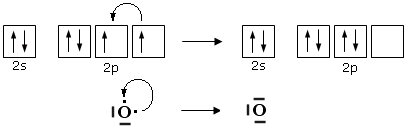
In questo modo l'ossigeno possiede ora un orbitale p vuoto che può utilizzare come accettore di un doppietto elettronico per formare un ulteriore legame chimico con lo zolfo di tipo dativo, per dare l’anidride solforosa SO2. Il passaggio dell’ossigeno ad una configurazione meno stabile richiede ovviamente energia, ma questa viene più che compensata dall’aumento di stabilità che si ottiene con la formazione di un ulteriore legame.
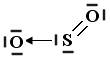
Nel caso lo Zolfo utilizzi entrambi i suoi doppietti solitari per formare due legami dativi con altrettanti atomi di ossigeno si forma l'anidride solforica SO3.
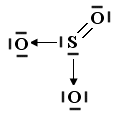
Il legame dativo ci permette di giustificare la capacità che hanno molti elementi ( in particolare gli elementi alla fine di un periodo) di formare un numero variabile di legami con l'ossigeno (valenza variabile) legandosi con esso in diverse proporzioni (legge delle proporzioni multiple di Dalton).
Il cloro, ad esempio, che possiede una configurazione superficiale s2 p5, presenta un elettrone spaiato e ben tre doppietti non condivisi disponibili per legami dativi. Si giustificano in tal modo l'esistenza di ben quattro composti ossigenati del cloro: l'anidride ipoclorosa Cl2O, l'anidride clorosa Cl2O3, l'anidride clorica Cl2O5 e l'anidride perclorica Cl2O7.
Non è necessario che un atomo liberi un orbitale per poter effettuare un legame dativo. In molti casi esistono già orbitali naturalmente liberi.
Un esempio si ha nella reazione di dissociazione ionica dell’acqua in ioni H+ e ioni OH-. In realtà in soluzione non esistono ioni H+ liberi poiché essi usano il loro orbitale 1s vuoto per legarsi, tramite legame dativo, ad uno dei due doppietti solitari dell’ossigeno di una molecola d’acqua, con formazione di ioni ossonio (o idronio) H3O+.
H2Ōı→H+ = H3O+
Una volta che lo ione ossonio si è formato, i 3 atomi di idrogeno sono perfettamente equivalenti ed i tre legami covalenti che li legano all’ossigeno risultano indistinguibili.
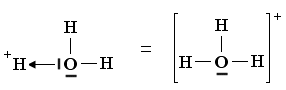
Ione ossonio
Promozione Elettronica
Come abbiamo appena visto, la formazione di legami stabilizza la molecola al punto che, in alcuni casi, un atomo può assumere configurazioni elettroniche meno stabili che tuttavia gli consentono di formare un maggior numero di legami. La promozione elettronica, ad esempio, è un processo di questo tipo, che consente ad un atomo di trasferire un elettrone da un orbitale superficiale saturo ad un orbitale superficiale vuoto. In questo modo un doppietto viene trasformato in due elettroni spaiati che, condivisi con altri atomi, possono essere utilizzati per formare due ulteriori legami chimici.
E’ il caso del Carbonio che, in quasi tutti i suoi composti promuove un elettrone dall’orbitale saturo 2s ad un orbitale 2p vuoto
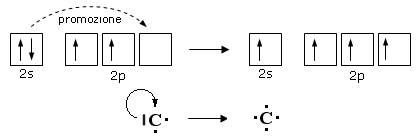
Pur passando da una configurazione elettronica superficiale più stabile ad una meno stabile, il Carbonio dispone ora di 4 elettroni spaiati (contro i due precedenti) che può condividere formando 4 legami chimici.
La promozione elettronica avviene quando la differenza di energia tra l’orbitale di partenza e quello di arrivo è piccola.
La geometria delle molecole: teoria VSEPR
I legami covalenti sono direzionali, nel senso che essi formano tra loro angoli caratteristici che determinano la geometria della molecola. La geometria di una molecola e di conseguenza gli angoli di legame possono essere previsti in modo semplice applicando la teoria VSEPR (Valence-Shell Electron-Pairs Repulsion = repulsione tra doppietti elettronici dello strato di valenza).
Secondo tale teoria i doppietti elettronici più esterni (strato di valenza), essendo carichi negativamente, si respingono, tendendo a disporsi il più lontano possibile gli uni dagli altri, in modo da rendere minima la forza repulsiva e più stabile l'intera molecola.
La teoria prevede inoltre che i doppietti solitari (non impegnati in legami) tendano ad occupare un volume maggiore rispetto ai doppietti elettronici condivisi (impegnati in legami) ed esercitino pertanto una forza repulsiva più intensa. In prima approssimazione possiamo stilare la seguente graduatoria relativa dell'intensità della repulsione esercitata tra coppie di elettroni
repulsione tra coppie solitarie > repulsione tra coppie solitarie e coppie di legame > repulsione tra coppie di legame
Inoltre nella teoria VSEPR i legami doppi e tripli vengono considerati alla stregua di legami semplici e la geometria di una molecola dipende unicamente dal numero di legami (indifferentemente semplici, doppi o tripli) e di coppie solitarie che presenta l’atomo centrale (numero sterico)
numero sterico = numero legami + numero coppie solitarie
La geometria di una molecola è determinata dal suo numero sterico (NS)
- NS=2 - Geometria lineare (AX2)
Molecole con due soli legami e nessun doppietto solitario (AX2) risultano lineari, con le coppie di legame che, respingendosi, si dispongono equidistanti, formando angoli di legame di 180°
X―A―X
Come abbiamo detto, i legami possono essere indifferentemente singoli, doppi o tripli. Presentano, ad esempio, geometria lineare l’idruro di Berillio (BH2), l’anidride carbonica (CO2) e l’acido Cianidrico (HCN)
H―Be―H O=C=O H―C ≡ N
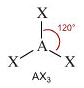
- NS=3 - Geometria trigonale planare (AX3, AX2E)
- Molecole con tre legami e nessun doppietto solitario (AX3) risultano trigonali planari, con le coppie di legame disposte equidistanti su di un piano, con angoli di legame di 120°. Presentano, ad esempio, geometria trigonale planare il cloruro di Boro (BCl3) e la formaldeide (H2CO).
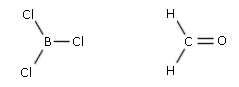
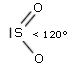
- Molecole con due legami ed un doppietto solitario (AX2E) risultano angolate, con un angolo di legame leggermente inferiore a 120° a causa della maggior repulsione del doppietto solitario sui doppietti di legame. Presenta una geometria angolata (derivata da una trigonale planare) l’anidride solforosa (SO2)
- NS=4 – Geometria tetraedrica (AX4, AX3E, AX2E2)
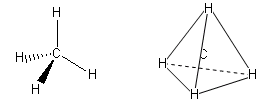
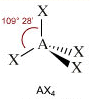
- Molecole con quattro legami e nessun doppietto solitario (AX4) risultano tetraedriche, con le coppie di legame disposte equidistanti ed angoli di legame di 109,5°. E’ il caso del metano (CH4). la cui molecola, come tutte le molecole tridimensionali, può essere rappresentata con legami a cuneo. Si utilizzano cunei pieni per rappresentare i legami che escono dal piano avvicinandosi all’osservatore e cunei tratteggiati per rappresentare i legami che si allontanano.
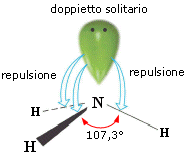
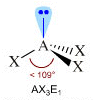
- Molecole con tre legami ed un doppietto solitario (AX3E) presentano una geometria piramidale di derivazione tetraedrica, con la coppia solitaria ad un vertice del tetraedro che comprime gli angoli di legame, portandoli ad un valore inferiore rispetto a quello caratteristico della geometria tetraedrica. E’ il caso dell’ammoniaca (NH3). la cui molecola piramidale presenta angoli di legame di circa 107°.
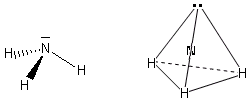
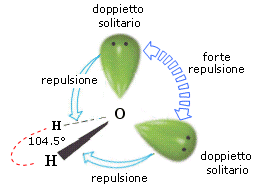
- Molecole con due legami e due doppietti solitari (AX2E2) presentano una geometria angolata di derivazione tetraedrica, con le due coppie solitarie ai due vertici del tetraedro che esercitano una forte repulsione e comprimono l’angolo di legame, portandolo ad un valore inferiore rispetto a quello caratteristico della geometria tetraedrica. E’ il caso dell’acqua (H2O). la cui molecola angolata presenta un angolo di legame di 104,5°.
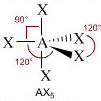
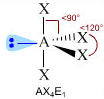
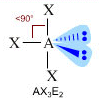
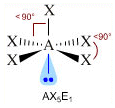
- NS=5 – Geometria bipiramidale trigonale (AX5, AX4E, AX3E2, AX2E3)
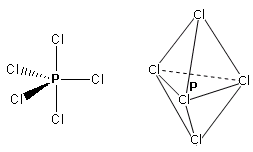
- Molecole con cinque legami e nessun doppietto solitario (AX5) risultano bipiramidali trigonali, con tre legami che si dispongono su di un piano (legami equatoriali) a 120° l’uno dall’altro e gli altri due legami (legami assiali) disposti perpendicolarmente, uno sopra e l’altro sotto al piano equatoriale, a formare due piramidi a base triangolare unite per la base. E’ il caso del Pentacloruro di Fosforo (PCl5).
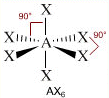
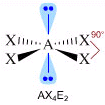
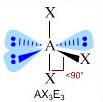
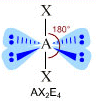
- NS=6 – Geometria ottaedrica (AX6, AX5E, AX4E2, AX3E3, AX2E4)
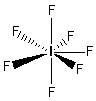
- Molecole con sei legami e nessun doppietto solitario (AX6) risultano ottaedriche con quattro legami equatoriali distanziati di 90° e due legami equatoriali. Presenta questa geometria l’Esafluoruro di Zolfo (SF6).
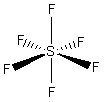
- NS=7 – Geometria bipiramidale pentagonale (AX7, AX6E, AX5E2, AX4E3, AX3E4, AX2E5)
- Molecole con sette legami e nessun doppietto solitario (AX7) risultano bipiramidali con cinque legami equatoriali distanziati di 72° e due legami equatoriali. Presenta questa geometria l’Eptafluoruro di Iodio (IF7).
Naturalmente in tutte le strutture, l’eventuale presenza di doppietti solitari modifica la geometria originaria, comprimendo gli angoli dei legami residui.
Geometrie VSEPR |
|||||
|
Coppie solitarie |
||||
0 |
1 |
2 |
3 |
4 |
|
NS=2 |
|
|
|
|
|
NS=3 |
|
|
|
|
|
NS=4 |
|
|
|
|
|
NS=5 |
|
|
|
|
|
NS=6 |
|
|
|
|
|
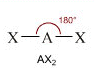
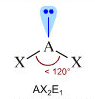
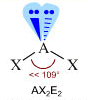
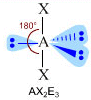
Legame covalente polare: elettronegatività e momento di dipolo
Quando gli elettroni vengono condivisi da atomi del medesimo elemento, ciascun atomo li attrae con la medesima intensità. In questo caso gli elettroni condivisi (elettroni di legame) possono essere immaginati come una nuvola negativa che si dispone in maniera uniforme e simmetrica intorno ai due nuclei senza produrre alcun tipo di polarità sulla molecola. Si parla in questo caso di legame covalente puro.
Nella maggior parte dei casi però gli atomi che formano il legame covalente appartengono ad elementi diversi che presentano una diversa forza di attrazione sugli elettroni di legame.
Si definisce elettronegatività χ (la lettera greca “chi”) la forza con cui un atomo attira a sé gli elettroni condivisi.
L'elettronegatività è una grandezza di difficile valutazione poiché, a differenza dell'affinità elettronica e dell'energia di ionizzazione che si riferiscono ad atomi isolati, l’elettronegatività si riferisce ad atomi legati ad altri atomi.
In generale il valore dell'elettronegatività può dunque variare, per uno stesso elemento, in relazione al tipo e al numero di atomi di altri elementi impegnati nel legame.
Nonostante ciò, al fine di avere a disposizione un parametro che permetta di valutare, anche se in modo approssimato, la polarità di un legame, sono stati proposti diversi metodi di calcolo per assegnare un valore di elettronegatività ai diversi elementi.
Tra i metodi più importanti vi sono quelli proposti da Mulliken e da Pauling.
L'elettronegatività secondo Mulliken è pari alla media aritmetica dell'energia di ionizzazione e dell'affinità elettronica.
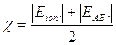
L'elettronegatività secondo Pauling di un elemento A viene calcolata conoscendo l'elettronegatività di un elemento B attraverso la seguente relazione
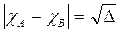
Pauling ammette cioè che la differenza di elettronegatività tra due elementi sia uguale alla radice quadrata di una quantità Δ, detta energia di risonanza ionico-covalente espressa in eV, il cui valore è dato da
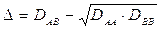
dove
DAB = energia di legame del composto A-B
DAA = energia di legame del composto A-A
DBB = energia di legame del composto B-B
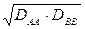 = media geometrica delle energie dei legami covalenti puri A-A e B-B, assunta come stima dell’energia di legame di un ipotetico legame covalente puro A-B
= media geometrica delle energie dei legami covalenti puri A-A e B-B, assunta come stima dell’energia di legame di un ipotetico legame covalente puro A-B
.
In altre parole, l'energia di risonanza ionico-covalente Δ misura la differenza di energia tra il legame covalente reale AB ed un ipotetico legame covalente puro AB.
Nel caso l'energia di legame sia espressa in kJ/mol o in kcal/mol è necessario applicare un coefficiente k di conversione (per trasformare in eV/particella), che vale rispettivamente 0,0103643 e 0,0433641.
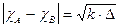
Per poter utilizzare la relazione di Pauling è evidentemente necessario fissare arbitrariamente l'elettron egatività di un elemento che faccia da riferimento. Pauling assunse per l'idrogeno χ = 2,1.
Esempio
Calcoliamo l'elettronegatività del Cloro secondo Pauling, sapendo che l'energia del legame H2 è 436 kJ/mol, del legame Cl2 è 242 kJ/mol e del legame HCl è 431 kJ/mol.
L'energia del legame HCl considerato come covalente puro è pari a 
L'energia di risonanza ionica-covalente è pari 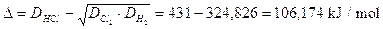
La differenza di elettronegatività calcolata è pertanto 
Sapendo che l'elettronegatività dell'idrogeno è convenzionalmente 2,1 si ottiene per il Cloro
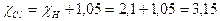
* * * * * * * * *
La scala di Mulliken è più rigorosa della scala di Pauling essendo costruita su grandezze misurabili. Nella pratica si usa però prevalentemente la scala di Pauling in quanto per molti elementi il valore dell'affinità elettronica è di difficile determinazione.
D'altra parte le due scale forniscono valori in gran parte coincidenti, risultando legate, anche se in modo approssimato, dalla seguente relazione
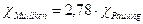
Esempio
Calcoliamo l'elettronegatività del cloro secondo Mulliken sapendo che la sua energia di ionizzazione è pari a 1260 kJ/mol(13,1 eV/particella) e la sua affinità elettronica è pari a -349 kJ/mol (-3,6 eV/particella) e convertiamo il valore ottenuto nella scala di Pauling
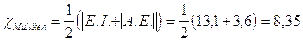
trasformiamo ora il valore nella scala di Pauling
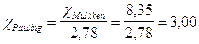
I valori di elettronegatività secondo Pauling si trovano tabulati nella tabella periodica e presentano il valore minimo in basso a sinistra (Francio = 0.7) e crescono diagonalmente fino ad assumere il valore massimo in alto a destra (Fluoro = 4).
Elettronegatività (Pauling) |
|||||||||||||||||
H |
|
He |
|||||||||||||||
Li |
Be |
|
B |
C |
N |
O |
F |
Ne |
|||||||||
Na |
Mg |
|
Al |
Si |
P |
S |
Cl |
Ar |
|||||||||
K |
Ca |
Sc |
Ti |
V |
Cr |
Mn |
Fe |
Co |
Ni |
Cu |
Zn |
Ga |
Ge |
As |
Se |
Br |
Kr |
Rb |
Sr |
Y |
Zr |
Nb |
Mo |
Tc |
Ru |
Rh |
Pd |
Ag |
Cd |
In |
Sn |
Sb |
Te |
I |
Xe |
Cs |
Ba |
La |
Hf |
Ta |
W |
Re |
Os |
Ir |
Pt |
Au |
Hg |
Tl |
Pb |
Bi |
Po |
At |
Rn |
Fr |
Ra |
Ac |
Rf |
Db |
Sg |
Bh |
Hs |
Mt |
Ds |
Rg |
Uub |
Uut |
Uuq |
Uup |
Uuh |
Uus |
Uuo |
Ce |
Pr |
Nd |
Pm |
Sm |
Eu |
Gd |
Tb |
Dy |
Ho |
Er |
Tm |
Yb |
Lu |
Th |
Pa |
U |
Np |
Pu |
Am |
Cm |
Bk |
Cf |
Es |
Fm |
Md |
No |
Lr |
E' evidente che tanto maggiore sarà la differenza di elettronegatività tra due elementi impegnati in un legame, tanto maggiore sarà la polarità del legame.
Dunque, quando si forma un legame covalente tra due atomi che presentano diversa elettronegatività la nube elettronica che costituisce il legame covalente risulta spostata verso l'atomo più elettronegativo. Quest'ultimo acquista pertanto una carica parzialmente negativa (indicata con -), mentre l'altro una carica parzialmente positiva (d+). La distribuzione asimmetrica della nuvola elettronica produce dunque due poli aventi carica opposta (dipòlo) ed il legame viene perciò definito covalente polare.
E’ ciò che accade, ad esempio nella molecola dell’acido Cloridrico (HCl) in cui l’Idrogeno condivide un elettrone con il Cloro. Il Cloro più elettronegativo presenta una parziale carica negativa e la molecola di HCl risulta polare
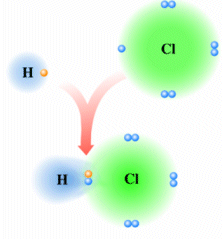
La distribuzione di carica elettrica di un dipolo può essere rappresentata tramite una mappa (o superficie) di potenziale elettrostatico (o densità elettronica) dove le tonalità del rosso indicano la carica negativa, quelle del blu la carica positiva, mentre il verde la neutralità.
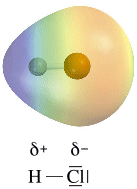
Maggiore è la differenza di elettronegatività (Δχ) tra i due elementi e maggiore sarà la polarità del legame (le cariche parziali saranno più vicine ad una intera carica).
Quando la differenza di elettronegatività tra i due elementi supera il valore critico di 1.9, si assume che l’elemento più elettronegativo sia in grado di strappare l’elettrone all’altro elemento ed il legame viene descritto come ionico. Possiamo dunque descrivere il legame ionico come un caso limite del legame covalente polare per Δχ > 1.9
L'intensità di un dipolo si esprime attraverso la determinazione del suo momento dipolare. Si definisce momento dipolare μ il prodotto della carica q associata ad uno dei baricentri di carica (la carica dell'altro baricentro ha valore uguale e di segno opposto) per la distanza r tra i baricentri.
μ = Q • r
L'unità di misura del momento dipolare è il debye (D).
Un momento dipolare presenta l'intensità di 1 debye quando 2 cariche elettriche di segno opposto, aventi intensità di 10-10 u.e.s. (unità elettrostatiche o franklin) si trovano alla distanza di 1 Å.
1 D = 10-10 ues Å = 3,335641·10-30 C m
Per caratterizzare la polarità di un legame covalente è possibile assegnargli una certa percentuale di carattere ionico, calcolabile in funzione del suo momento dipolare.
La percentuale di carattere ionico si calcola come rapporto percentuale tra il momento dipolare effettivo (misurato) ed il momento dipolare di un teorico legame ionico
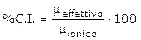
Il momento dipolare di un ipotetico legame completamente ionico si calcola con
μionico = Qe• r = 4,8 • r
dove Qe è la carica dell'elettrone (e = 4,8 10-10 u.e.s.) ed r il valore della lunghezza del legame in Å.
Ad esempio, sapendo che il momento dipolare dell’acido cloridrico è μHCl = 1,1 D e la lunghezza del legame H-Cl è di 1,27 Å, si calcola
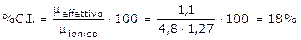
Sopra l’atomo di Cloro è presente una parziale carica negativa (d-) pari al 18% dell’intera carica dell’elettrone, mentre sopra l’atomo di Idrogeno sarà presente una parziale carica positiva (d+) della medesima intensità.
La polarità di un legame può anche essere stimata utilizzando la relazione di Pauling che correla la percentuale di carattere ionico alla differenza di elettronegatività (Δχ).
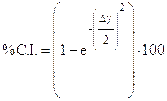
Ad esempio, sapendo che la differenza di elettronegatività tra Idrogeno e Fluoro è pari a Δχ = χF – χH = 4 - 2,2 = 1,8, possiamo stimare la percentuale di carattere ionico dell’acido fluoridrico
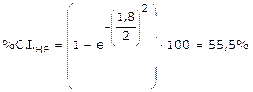
Il momento dipolare è una grandezza vettoriale, che viene rappresentata con una freccia orientata dal polo positivo al quello negativo.
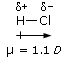
Nelle molecole in cui sono presenti più legami il momento di dipolo dell’intera molecola risulta essere la somma vettoriale dei momenti di dipolo dei singoli legami. Se il momento di dipolo risultante è diverso da zero allora la molecola è polare. Se il momento di dipolo risultante è uguale a zero, la molecola risulta apolare (anche se i suoi legami sono polari).
La polarità di una molecola dipende quindi non solo dalla polarità dei suoi legami, ma anche dalla sua geometria.
Così, ad esempio, se confrontiamo la polarità dell’anidride carbonica e dell’acqua, troveremo che mentre l’anidride carbonica è apolare, l’acqua è polare. L’anidride carbonica è infatti una molecola lineare ed il momento di dipolo dei suoi due legami risulta essere uguale e contrario, per cui il momento risultante è nullo.
L’acqua è invece una molecola angolata e la polarità dei suoi due legami si compone vettorialmente per dare un momento di dipolo diverso da zero. L’acqua è un dipolo.
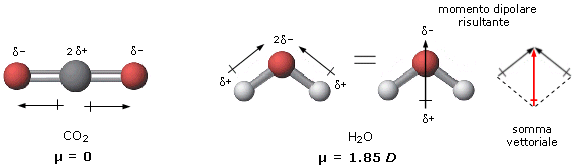
In effetti il momento dipolare totale di una molecola è dato dalla somma vettoriale non solo dei momenti dipolari relativi ai legami covalenti, ma anche ai dipoli associati alle coppie solitarie presenti nella molecola.
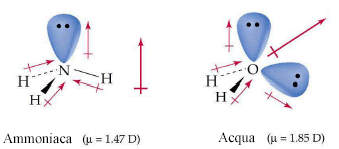
Una molecola polare è un minuscolo dipòlo il quale è in grado di ruotare orientandosi opportunamente se posto in un campo elettrico.
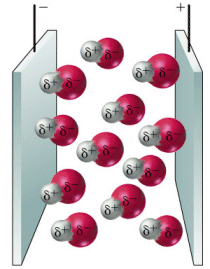
Dipoli orientati in un campo elettrico
Molecole con elettroni spaiati e paramagnetismo
Non sempre gli atomi utilizzano tutti i loro elettroni spaiati per effettuare legami chimici. In qualche caso può accadere che in una molecola sopravvivano degli orbitali insaturi.
Un tipico esempio di tale comportamento è rappresentato dal monossido di azoto.
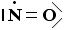
Tutte le sostanze che si trovano a possedere un elettrone spaiato risultano essere paramagnetiche, vengono cioè debolmente attratte dai poli di un magnete. Tale comportamento è dovuto proprio al debole campo magnetico associato all'elettrone, non compensato in questo caso da un elettrone con spin opposto.
Strutture di Lewis molecolari e carica formale
La convenzione introdotta da Lewis per rappresentare gli elettroni di valenza degli elementi viene utilizzata anche per rappresentare intere molecole. Per scrivere la struttura di Lewis di una molecola è necessario conoscere la sua formula molecolare e la connettività.
La formula molecolare e la connettività sono determinate sperimentalmente e devono essere note. La connettività o costituzione è l’ordine con cui gli atomi di una molecola sono connessi.
Ad esempio il nitrito di metile ha formula molecolare CH3NO2 e la sua connettività è C-O-N-O con tutti gli idrogeni legati al carbonio.
Vediamo di seguito i 6 passaggi necessari per scrivere correttamente una struttura di Lewis molecolare. Useremo il nitrito di metile CH3NO2.
- Determinare il numero degli elettroni di valenza della molecola.
Per una molecola neutra, il numero di elettroni di valenza è uguale alla somma degli elettroni di valenza degli atomi coinvolti (elettroni superficiali che compaiono nella struttura di Lewis dell’elemento). Si ricordi che il numero di elettroni superficiali coincide in genere con il numero d’ordine del gruppo chimico al quale l’elemento appartiene. Così l’Ossigeno (VI gruppo A) possiede 6 elettroni di valenza, l’Azoto (V gruppo A) ne possiede 5, il Carbonio 4 (IV gruppo A) e l’Idrogeno 1 elettrone di valenza (I gruppo A).
Per una molecola elettricamente carica il numero di elettroni di valenza è uguale al numero di elettroni di valenza degli atomi, al quale va sommato il numero delle cariche negative o sottratto il numero delle cariche positive. Ad esempio l’anione solfato SO42- possiede 32 elettroni di valenza (6 per l’atomo di Zolfo, 6 per ogni atomo di ossigeno e 2 per le due cariche negative dell’anione).
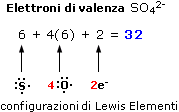
Il nitrito di metile (CH3NO2) presenta 24 elettroni di valenza. Ogni idrogeno contribuisce infatti con 1 elettrone di valenza, il carbonio con 4, l’azoto con 5 ed ogni atomo di ossigeno con 6 per un totale di 24 elettroni.
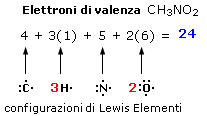
- Costruire un primo schema di legame, rispettando la connettività e collegando gli atomi con un legame covalente semplice.
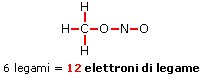
- Determinare gli elettroni residui da posizionare. Sottrarre gli elettroni di legame dagli elettroni di valenza, ottenendo in tal modo il numero di elettroni che devono ancora essere posizionati. Nel nitrito di metile CH3NO2, gli elettroni di legame sono 12, mentre quelli di valenza sono 24 e devono pertanto essere ancora posizionati 24 – 12 = 12 elettroni.
- Aggiungere gli elettroni residui come coppie di elettroni di non-legame (elettroni non condivisi o coppie solitarie o lone pairs) in modo che il maggior numero di atomi presenti 8 elettroni (ovviamente non l’idrogeno), iniziando con gli atomi più elettronegativi.
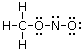
- Spostare coppie solitarie per completare l’ottetto. Se un atomo non ha l’ottetto completo, usare una coppia di elettroni solitari dell’atomo adiacente che possiede il maggior numero di doppietti non condivisi, per formare un doppio o un triplo legame. Nella struttura precedente si osserva, ad esempio, che l’azoto ha solo 6 elettroni (4 condivisi con i due atomi di Ossigeno e 2 non condivisi). Trasferiamo dunque un doppietto solitario dall’Ossigeno terminale (che ha 3 doppietti non condivisi, contro i 2 doppietti dell’ossigeno centrale), per formare un doppio legame N=O.
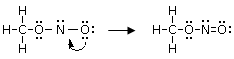
La regola per la quale ogni atomo legato deve avere non più di 8 elettroni superficiali (regola dell’ottetto) vale rigorosamente solo per gli elementi non metallici del secondo periodo chimico (C N O F) i quali, non possedendo orbitali d, non sono in grado di trasferirvi elettroni (promozione elettronica) per aumentare il numero dei loro legami. I primi elementi del secondo periodo possono avere meno di otto elettroni nel loro stato legato (4 per il Berillio e 6 per il Boro = ottetto incompleto). Gli elementi non metallici dei periodi superiori al secondo possono invece avere più di 8 elettroni superficiali (ottetto espanso) nel loro stato legato. In particolare quelli del gruppo V-A, come il Fosforo, possono avere 10 elettroni superficiali, mentre quelli del gruppo VI-A come lo Zolfo possono arrivare a 12 e quelli del gruppo VII-A, come il Cloro ne possono avere 14.
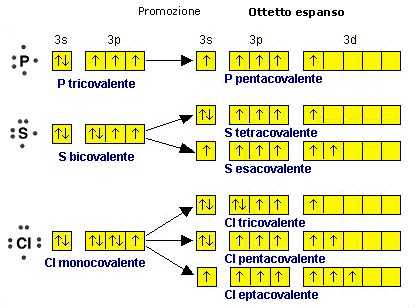
Ad esempio Lo Zolfo completa l’ottetto nell’acido solfidrico, mentre arriva a 10 elettroni nell’acido solforoso e a 12 elettroni nell’acido solforico
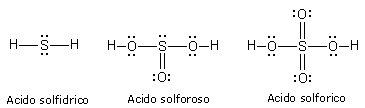
- Calcolare la carica formale di ciascun atomo. La carica atomica formale viene definita come la carica che un atomo avrebbe se tutti i suoi legami venissero considerati come covalenti puri. La carica formale di ciascun atomo e' data dunque dalla differenza tra gli elettroni di valenza dell'atomo isolato e gli elettroni di valenza dell'atomo legato nella molecola. Per determinare gli elettroni di valenza dell’atomo legato (elettroni che l’atomo possiede nella molecola) è necessario assegnargli un elettrone per ogni coppia di elettroni di legame ed entrambi gli elettroni di ogni sua coppia solitaria. Si confrontano poi gli elettroni di valenza dell’atomo legato con gli elettroni di valenza dell’atomo isolato neutro. Ogni elettrone in eccesso rappresenta una carica formale negativa. Ogni elettrone in difetto una carica formale positiva.
carica formale = elettroni di valenza atomo isolato – elettroni di valenza atomo legato
carica formale = elettroni di valenza – [½ elettroni di legame + elettroni solitari]
Naturalmente la somma delle cariche formali di tutti gli atomi di una molecola o ione deve essere uguale alla carica elettrica complessiva della molecola.
Per calcolare la carica formale sostituiamo i trattini che rappresentano i legami con coppie di puntini (i due elettroni condivisi) e successivamente contiamo gli elettroni intorno ai singoli atomi confrontandoli con i rispettivi elettroni di valenza degli atomi isolati
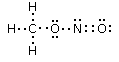
Intorno a ciascun atomo di ossigeno vi sono 6 elettroni. Poiché l’atomo di ossigeno isolato possiede 6 elettroni superficiali, i due atomi di ossigeno presentano carica formale nulla (6-6=0)
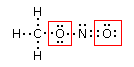
Intorno all’atomo di carbonio vi sono 4 elettroni, mentre intorno all’atomo di azoto vi sono 5 elettroni. Poiché gli atomi di carbonio e di azoto isolati possiedono rispettivamente 4 e 5 elettroni superficiali, la loro carica formale è nulla.
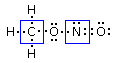
Esaminiamo ora il nitrometano, un composto che presenta la medesima formula molecolare del nitrito di metile CH3NO2, ma diversa connettività o costituzione (nitrometano e nitrito di metile sono due isomeri costituzionali). Nel nitrometano il carbonio si lega direttamente al’azoto il quale si lega ai due atomi di ossigeno. Il primo schema di legame è dunque il seguente
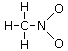
Abbiamo posizionato 6 legami per un totale di 12 elettroni. Gli elettroni di valenza sono sempre 24, come per il nitrito di metile (gli atomi sono gli stessi) e ci rimangono dunque altri 12 elettroni da posizionare. Assegnamo 3 doppietti solitari a ciascun atomo di ossigeno (più elettronegativo dell’azoto) in modo da raggiungere una configurazione ad 8 elettroni
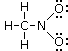
In questo modo l’azoto non presenta tuttavia l’ottetto completo (ha solo 3 legami per un totale di 6 elettroni) e quindi trasferiamo un doppietto solitario di uno dei due atomi di Ossigeno per formare un doppio legame N=O
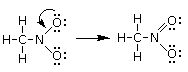
Calcoliamo ora la carica formale di ciascun atomo. Sostituiamo i trattini che rappresentano i legami con coppie di puntini (i due elettroni condivisi) e successivamente contiamo gli elettroni intorno ai singoli atomi (elettroni di valenza degli atomi legati)
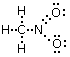
L’atomo di ossigeno con il doppio legame presenta 6 elettroni, di cui 2 elettroni per i due legami e 4 per i due doppietti solitari). Avendo l’ossigeno isolato 6 elettroni di valenza la sua carica formale è nulla. L’atomo di ossigeno con il legame semplice presenta invece 7 elettroni, di cui 1 elettrone per il legame e 6 per i tre doppietti solitari. Avendo dunque un elettrone in più rispetto ai 6 elettroni di valenza di un atomo di ossigeno isolato, la sua carica formale è -1.

L’atomo di Azoto presenta 4 elettroni (metà degli 8 elettroni che formano i suoi 4 legami) Avendo dunque un elettrone in meno rispetto ai 5 elettroni di valenza di un atomo di Azoto isolato, la sua carica formale è +1.

L’atomo di Carbonio presenta 4 elettroni (metà degli 8 elettroni che formano i suoi 4 legami) Avendo dunque il medesimo numero di elettroni di un atomo di carbonio isolato, la sua carica formale è nulla.

La formula di Lewis del nitrometano sarà pertanto
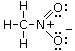
Si rammenti che una formula di Lewis non è completa se non presenta le corrette cariche formali.
Il Legame covalente: Teoria del legame di valenza (VB)
La teoria del legame di valenza (Valence Bond Theory) fu proposta nel 1927 da W.Heitler e F.London e successivamente ampliata e sviluppata da L.Pauling con l’introduzione dei concetti di risonanza (1928) e di ibridazione orbitalica (1930). La teoria interpreta la formazione del legame covalente mediante il concetto quantomeccanico di orbitale.
Il legame covalente, che nella teoria di Lewis viene visto come una condivisione da parte di due atomi di una coppia di elettroni, viene descritto come una sovrapposizione degli orbitali atomici che ospitano i due elettroni spaiati da condividere.
Le funzioni d’onda dei due orbitali si sommano (in modo analogo ai fenomeni di interferenza per le onde meccaniche) per dare una nuova funzione d’onda che descrive un nuovo orbitale.
Il nuovo orbitale appartiene ad entrambi gli atomi legati ed ospita i due elettroni con spin antiparallelo.
Nel caso del legame covalente semplice che tiene uniti due atomi di Idrogeno nella molecola H2, ad esempio, abbiamo una sovrapposizione di due orbitali 1s. Se indichiamo i due atomi di Idrogeno con HA e HB, le due funzioni d’onda che si sommano sono ΨA(1s) e ΨB(1s)
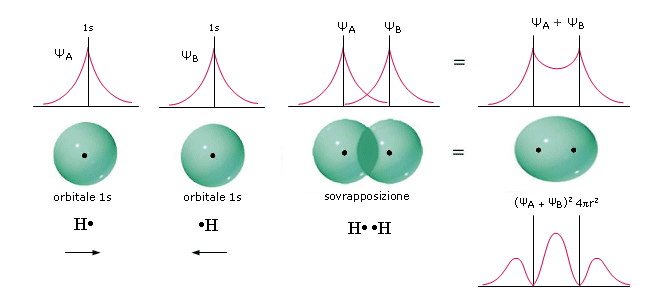
La funzione di distribuzione radiale della densità elettronica (probabilità) del nuovo orbitale che si è formato mostra un massimo tra i due nuclei. Si suppone che, quando gli atomi di H si avvicinano, ciascun elettrone condiviso possa passare da un nucleo all’altro, cioè che a distanze ravvicinate i nuclei non ‘distinguano’ gli elettroni di legame.
Nel formare i legami gli orbitali, se possibile, tendono a massimizzare la regione di sovrapposizione. Gli orbitali di tipo p, ad esempio, tendono a sovrapporsi lungo il loro asse maggiore. Nella molecola biatomica del Fluoro F2, ad esempio, due orbitali 2p si sovrappongono lungo l’asse maggiore, utilizzando i lobi aventi il medesimo segno, in modo che la funzione d’onda tra i due nuclei si rinforzi, aumentando la densità elettronica.

Questo tipo di sovrapposizione genera un legame covalente particolarmente intenso, detto legame σ.
Nel caso di legami covalenti doppi e tripli, solo una coppia di orbitali p può generare un legame σ. Gli altri orbitali p, essendo disposti uno perpendicolarmente all’altro, sono costretti a sovrapporsi lateralmente (lungo l’asse minore). Questo tipo di legame covalente è più debole (a causa della minor sovrapposizione) ed è detto legame π.
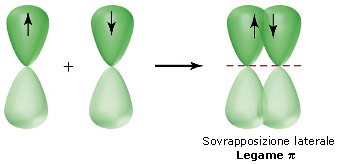
Quando in una molecola si forma un legame covalente doppio si genera un legame σ lungo la congiungente i due nuclei ed un legame π costituito da due nuvole elettroniche disposte simmetricamente (sopra e sotto) rispetto al legame σ. Un doppio legame è una struttura rigida e non consente la libera rotazione dei due atomi legati attorno all’asse di legame.
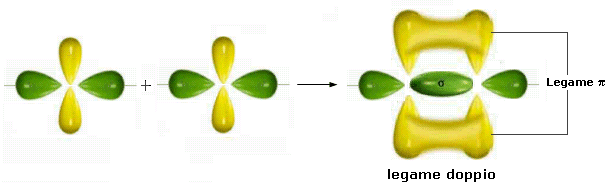
Il legame doppio è quindi più forte di un legame semplice, ma presenta tuttavia una forza inferiore a quella di due legami semplici essendo costituito da un legame σ (più forte) ed un legame π (più debole).
Quando in una molecola si forma un legame covalente triplo si genera un legame σ lungo la congiungente i due nuclei e due legami π costituiti da quattro nuvole elettroniche disposte simmetricamente ai quattro lati del legame σ (un legame sopra-sotto ed un legame davanti-dietro). Anche un triplo legame è una struttura rigida e non consente la libera rotazione dei due atomi legati attorno all’asse di legame.
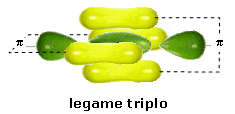
In una molecola biatomica come l’azoto (N2), ad esempio, in cui due atomi di Azoto sovrappongono tre coppie di orbitali p formando un legame covalente triplo, gli orbitali px si compenetrano lungo la congiungente i due nuclei formando un legame di tipo σ, mentre gli altri orbitali p si sovrappongono lateralmente dando origine a due legamiπ che presentano un massimo di densità elettronica sopra e sotto l’asse internucleare.
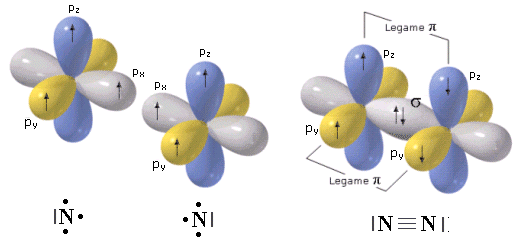
Il legame triplo è quindi più forte di un legame semplice, ma presenta tuttavia una forza inferiore a quella di tre legami semplici essendo costituito da un legame σ (più forte) e da due legami π (più deboli).
In definitiva, nel caso in cui la densità elettronica si concentri sull’asse internucleare, si parla di legameσ, nel caso si concentri sopra e sotto l’asse internucleare si parla di legameπ.
I legami σ presentano una simmetria cilindrica e sono quindi invarianti per rotazione attorno all’asse di legame.
I legami π non sono cilindricamente simmetrici rispetto all'asse di legame, poiché la funzione d’onda cambia di segno per rotazione attorno all’asse.
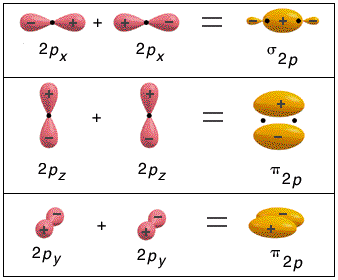
Presentano simmetria σ anche i legami che si formano per sovrapposizione di due orbitali s, come nella molecola H2, o per sovrapposizione di un orbitale s con un orbitale p, come nella molecola HCl.

Ibridazione orbitalica
Per formare legami, gli atomi possono ricombinare gli orbitali atomici (s,p,d) per dar luogo ad un ugual numero di orbitali atomici detti orbitali ibridi. Questo processo, detto ibridazione, è un procedimento di combinazione matematica delle funzioni d’onda originarie.
L’ibridazione interessa orbitali superficiali (di valenza) con contenuto energetico non molto diverso. Gli orbitali ibridi così formati sono tutti di uguale forma ed energia e sono orientati in modo da interferire il meno possibile fra di loro, massimizzando la reciproca distanza.
Gli orbitali ibridi più importanti sono quelli che si formano dalla combinazione di un orbitale s con uno o più orbitali p. La superficie di contorno di tali orbitali ibridi è costituita da due lobi contrapposti di diversa dimensione in cui la funzione d’onda Ψ assume segno opposto. Il lobo di dimensione maggiore è quello che viene utilizzato nei legami.
La combinazione di un orbitale di tipo s e uno di tipo p dà origine a due orbitali ibridi detti orbitali sp
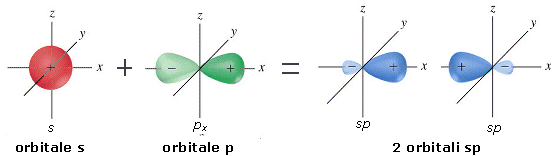
Gli orbitali sp si dispongono a 180° l’uno rispetto all’altro. Nella visione d’insieme spesso si omette di rappresentare il lobo minore di ciascun orbitale ibrido.
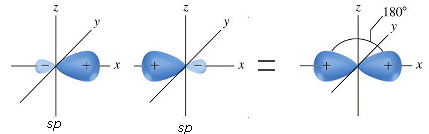
Gli orbitali p non ibridati si orientano perpendicolarmente alla retta di ibridazione e perpendicolarmente tra loro.
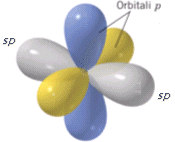
L’ibridazione sp è tipica di molecole con una geometria lineare. Presentano un’ibridazione sp l’atomo di Berillio nell’idruro di Berillio (BH2), l’atomo di carbonio nell’anidride carbonica (CO2) e gli atomi di carbonio uniti da un legame covalente triplo (-C≡C-) come ad esempio nella molecola dell’etino HC≡CH
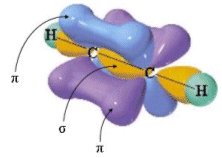
Etino
Va detto che, per semplicità, si disegnano normalmente gli ibridi con il piano nodale passante per il nucleo, mentre il nucleo si trova in una zona a densità elettronica non nulla.
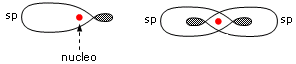
La combinazione di un orbitale di tipo s e di due orbitali di tipo p dà origine a tre orbitali ibridi detti orbitali sp2 che si dispongono su di un piano a 120° l’uno dall’altro.
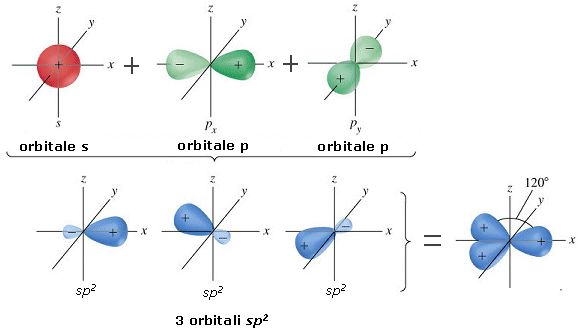
L’orbitale p non ibridato si dispone perpendicolarmente al piano di ibridazione.
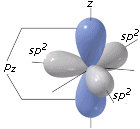
L’ibridazione sp2 è tipica di molecole con una geometria trigonale planare. Presentano un’ibridazione sp2 l’atomo di Boro nel cloruro di Boro (BCl3) e gli atomi di carbonio uniti da un legame covalente doppio (>C=C<), come ad esempio nella molecola dell’etene (o etilene) H2C=CH2
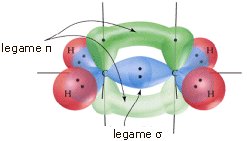
Etene
La combinazione di un orbitale di tipo s e di tre orbitali di tipo p dà origine a quattro orbitali ibridi detti orbitali sp3 che puntano verso i vertici di un tetraedro, disponendosi a 109,5° l’uno dall’altro.
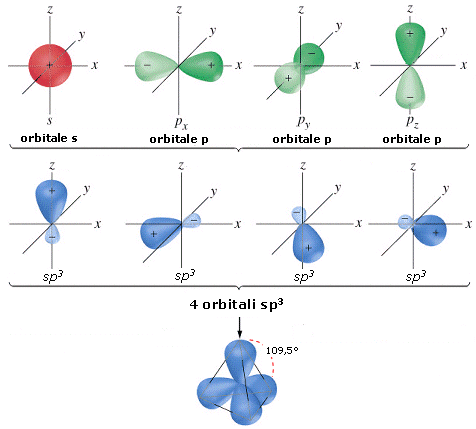
L’ibridazione sp3 è tipica di molecole con una geometria tetraedrica. Presenta un’ibridazione sp3 l’atomo di Carbonio nel metano (CH4) ed in tutti i casi in cui forma quattro legami covalenti semplici.
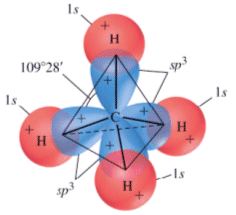
Metano
Sono possibili ibridazioni più complesse che coinvolgono anche gli orbidali d e che corrispondono alle geometrie già studiate con la teoria VSEPR.
Ibridazione |
Geometria |
Molecola |
sp |
lineare |
|
sp2 |
Trigonale planare |
|
sp3 |
Tetraedrica |
|
sp3d |
Bipiramidale trigonale |
|
sp3d2 |
Ottaedrica |
|
sp3d3 |
Bipiramidale pentagonale |
|




fonte: http://www.fileden.com/files/2008/11/5/2174401/Chimica.zip
fonte: http://rodomontano.altervista.org/chimica.php
sito web: http://rodomontano.altervista.org/
Autore del testo: Rodomontano
- Acidi
- Acidi e basi
- Acidi nucleici
- Acido folico e sulfamidici
- Acido salicilico
- Agenti chimici
- Aluminium Eng vers
- Analisi batteriologica acqua
- Appunti curiosi
- Biochimica
Biochimica industriale appunti
- Biochimica quiz
- Biocombustibili e biodiesel
- Chimica analitica glossario
- Chimica appunti
- Chimica appunti parte 1
- Chimica appunti parte 2
- Chimica appunti parte 3
- Chimica quiz
- Chimica e alchimia
- Chimica e gli alimenti
Chimica elementi gruppo 15
Chimica elementi gruppo 16
- Chimica farmaceutica
Chimica fotografia B&W
- Chimica generale
- Chimica generale 2
- Chimica generale 3
- Chimica generale 4
- Chimica generale 5
- Chimica generale 6
- Chimica generale 7
- Chimica generale 8
- Chimica generale 9
- Chimica generale 10
- Chimica generale appunti
- Chimica inorganica
- Chimica in versi
- Chimica in versi parte 2
Chimica peso molare
- Chimica misure e grandezze
- Chimica organica
- Chimica riassunto
- Chimica riassunti
- Chimica tabelle utili
- Chimica termini tecnici A
- Chimica termini tecnici B
- Chimica termini tecnici C
- Chimica termini tecnici D
- Chimica termini tecnici E
- Chimica termini tecnici F
- Chimica termini tecnici G H
- Chimica termini tecnici I
- Chimica termini tecnici L M
- Classific. e nomenclatura
- Cobalto e le sue leghe
- Coenzima q10 proprietà
- Composti organici biomole..
- Elementi chimici
- Elettrochimica
- Elettrolisi
- Esperimenti di chimica
- Esperimenti di chimica
- Glucidi riassunto
- Idrogeno caratteristiche
- Legami chimici
- Meccanica ondulatoria
- Microscopio di Heisenberg
- Modelli atomici
- Modelli atomici quanto-mec
- Monossido di carbonio
- Nomenclatura acidi ternari
- Nomenclatura idruri
- Numeri di ossidazione
- Osmosi inversa e idrologia
- Peso atomico azoto
- Peso atomico carbonio
- Peso atomico cloro
- Peso atomico ossigeno
- Peso atomico sodio
- Peso atomico zolfo
- Probabilità quantistica
- Quiz di chimica
- Riconoscimento gruppi
- Sali binari e sali ternari
- Storia della chimica
- Storia origini chimica
- Struttura atomica
- Struttura atomica 2
- Tabella punti di fusione
- Tavola periodica elementi
- Tavola periodica 2
- Valenza
Chimica appunti parte 3
Visita la nostra pagina principale
Chimica appunti parte 3
Termini d' uso e privacy